
Гемофагоцитарный синдром связанный с инфекцией
В статье рассмотрены вопросы патогенеза гемофагоцитарного синдрома (ГФС), связь с инфекциями, а также классификация и дифференциально-диагностические признаки. Представлен случай ГФС у ребенка с септикопиемической формой сальмонеллеза, описаны клинически
The article concentrates upon the issues of hemophagocytic syndrome (HFS) pathogenesis, its connection with infections, as well as its classification and differential-diagnostic signs. A clinical case of HFS in a child with septicopyemic form of salmanellosis was presented, clinical symptoms and laboratory data of the patient were described.
ГФС может быть первичным (наследственным вследствие генетического дефекта функции макрофагов) и вторичным или приобретенным, ассоциированным с инфекционным заболеванием, опухолями, аутоиммунными болезнями, наследственными болезнями обмена. При классических наследственных формах ГФС дети в силу особенностей клинической манифестации нередко получают терапию в отделениях реанимации и интенсивной терапии инфекционных стационаров с такими диагнозами, как сепсис или внутриутробная генерализованная инфекция, и часто истинный диагноз устанавливается посмертно. С другой стороны, банальные инфекции вирусной или бактериальной этиологии могут осложняться развитием жизнеугрожающего ГФС, требующего ранней диагностики для своевременного начала не только этиотропной, но и иммуносупрессивной терапии для подавления чрезмерной активации иммунного ответа [1–3].
Для постановки клинического диагноза ГФC необходимо наличие большинства из нижеперечисленных признаков:
- упорная фебрильная лихорадка более 7 дней, не купирующаяся на фоне антибактериальной или противовирусной терапии;
- гепатоспленомегалия с признаками функциональной недостаточности печени;
- геморрагический синдром, обусловленный как коагулопатией за счет ДВС-синдрома, так и печеночной недостаточностью;
- желтуха;
- отеки;
- лимфаденопатия;
- неврологические симптомы: рвота, отказ от еды, судороги, менингеальные знаки с повышением уровня белка и лейкоцитов в ликворе;
- панцитопения;
- гипертриглицеридемия, гипоальбуминемия, гипонатриемия, повышение уровня билирубина, лактатдегидрогеназы;
- повышение уровня ферритина в сыворотке крови;
- низкая активность натуральных киллеров (NK-клеток);
- высокое содержание провоспалительных цитокинов в крови и ЦНС, особенно уровня растворимого рецептора ИЛ-2 (СD25);
- в миелограмме полиморфная картина с активированными моноцитами/макрофагами, явления фагоцитоза клеточных элементов (эритроцитов, реже лейкоцитов и тромбоцитов).
Прогноз при развитии ГФC часто неблагоприятный с летальным исходом у детей преимущественно дошкольного возраста. В протоколы лечения первичного ГФC внесены глюкокортикостероиды (дексаметазон) и цитостатические препараты (этопозид, циклоспорин А) для подавления провоспалительной активности фагоцитирующих клеток с последующей аллогенной трансплантацией стволовых клеток. Елиного подхода к терапии ГФC, ассоциированного с инфекционным заболеванием, не существует. Этиотропной терапии недостаточно для купирования ГФC, а иммуносупрессивная терапия может оказать отрицательное влияние на динамику инфекционного процесса. Показано введение высокодозного иммуноглобулина в дозе 1–2 мг на кг массы тела в сутки на курс лечения. Плазмоферез целесообразно включать в комплекс средств патогенетической терапии для контроля за гиперцитокинемией [2, 4].
В доступной литературе мы не встретили описаний течения сальмонеллеза, осложненного развитием ГФC у детей.
Представляем клиническое наблюдение злокачественного течения у ребенка 3 лет генерализованной инфекции в виде септикопиемического варианта сальмонеллеза с наличием гнойных очагов инфекции в селезенке, брыжеечных лимфоузлах, осложненной развитием гемофагоцитарного синдрома и полиорганной недостаточностью (печеночной, энтеральной, дыхательной, сердечно-сосудистой, ДВС-синдромом с множественными кровоизлияниями во внутренние органы, панцитопенией). Подобное течение сальмонеллеза является редко встречающимся в отличие от локализованных форм, мало описано как в отечественной, так и в зарубежной литературе. В практике гемофагоцитарный синдром (ГФС), проявляющийся лихорадкой, полиорганной патологией с развитием недостаточности вовлеченных в процесс органов (чаще печени) и обязательным гемофагоцитозом (поглощением фагоцитами форменных элементов крови), представляет серьезную проблему.
Мальчик С., 3 года, родился 19.01.09 г., поступил в педиатрическое отделение центральной районной больницы Новосибирской области 27.08.12 г. с жалобами на повышение температуры тела до 39,6 °С, общую слабость, вялость, рвоту, боли в животе, чаще в области пупка, не связанные с приемом пищи, отсутствие аппетита.
Из анамнеза жизни известно, что мальчик родился от первой беременности, роды кесаревым сечением, с массой тела 2900 г и длиной 49 см. Грудное вскармливание — до 1 мес. В роддоме привит БЦЖ, рубчик имеется. В дальнейшем прививался по календарю. На первом году жизни перенес пневмонию. На втором и третьем году — нечастые ОРЗ (до 3 раз за год), 1 раз — лакунарная ангина. В 3 года перелом левого бедра.
Из анамнеза последнего заболевания известно, что ребенок заболел остро 22.08.12 (за 5 дней до поступления в стационар). Дебют заболевания с подъема температуры до фебрильных цифр, катаральных явлений в носоглотке. Со вторых суток заболевания отмечалась повторная рвота. Мама лечила ребенка самостоятельно (цефазолин, Нурофен, Цефекон симптоматически в возрастных дозах). Обращение к педиатру 27.08.12, который обнаружил увеличение живота в объеме и его болезненность при пальпации, плотную консистенцию печени и увеличение ее размеров (выстояла из подреберья на 8 см), а также селезенки (на 7 см). Диарейный синдром в анамнезе заболевания отсутствовал.
Госпитализирован в ЦРБ в тяжелом состоянии, температура к 6-му дню болезни (моменту госпитализации) была нормальной, но при этом выявлены признаки эндотоксикоза — вялость, анорексия, бледно-желтый цвет кожного покрова, вздутие живота, выраженная гепатоспленомегалия, признаки нарушения питания (масса тела 14 кг вместо положенных 16 кг, гипопротеинемия, гипонатриемия), признаки печеночной недостаточности (наряду с гепатомегалией непрямая гипербилирубинемия (29,3 мкмоль/л), гипопротеинемия (49 г/л), гиперферментемия (аспартатаминотрансфераза (АСТ) — 366 ед/л, аланинаминотрансфераза (АЛТ) — 174 ед/л), признаки глубокой гипокоагуляции. Установлена панцитопения (лейкоциты 2,8 × 10 9 /л, на следующие сутки 0,8 × 10 9 /л; эритроциты — 3,5 × 10 12 /л и 2,8 × 10 12 /л соответственно, гемоглобин 90 и 67 г/л, тромбоциты — 124 × 10 9 /л и 91 × 10 9 /л). В формуле крови на фоне лимфоцитоза отмечалось резкое преобладание юных форм (палочкоядерные — 17%), что является одним из маркеров сепсиса. За 17 часов пребывания ребенка в ЦРБ стула не было. Основным диагнозом выставлена генерализованная вирусно-бактериальная инфекция, тяжелая форма. Сепсис на этапе лечения в ЦРБ был внесен в перечень предполагаемых диагнозов.
Проводимое лечение: антибактериальная терапия Тиенамом, дезинтоксикационная и гемостатическая (глюкозо-солевые растворы, Волювен, плазма, Викасол, сорбенты, пробиотики).
Через 17 часов ребенок был переведен в больницу Новосибирска (отделение реанимации и интенсивной терапии — ОРИТ). При поступлении состояние ребенка расценено как тяжелое, обусловленное токсикозом, панцитопенией, синдромом полиорганной недостаточности (СПОН) с преобладанием недостаточности функции печени (повышение трансаминаз в 10 раз, гипокоагуляция, гипопротеинемия).
В слюне методом полимеразной цепной реакции (ПЦР) обнаружена ДНК вируса Эпштейна–Барр. Данные УЗИ органов брюшной полости: увеличение размеров печени (правая доля 130 мм, левая доля 80 мм) и селезенки (98 × 43 см), утолщение стенки желчного пузыря, диффузные изменения в печени и почках, свободная жидкость в брюшной полости, правой плевральной полости.
Данные рентгеновской компьютерной томографии (КТ) головного мозга: КТ-признаков патологических изменений вещества мозга не выявлено, исключены отек мозга, кровоизлияние в мозг. Левосторонний гайморит. Маркеры вирусных гепатитов, ВИЧ-инфекции не обнаружены.
Через 6 часов после поступления резкое ухудшение состояния: профузное кровотечение из пищевода, желудка с развитием гемической гипоксии (падение гемоглобина до 43 г/л). По показателям гемостаза признаки глубокой гипокоагуляции (протромбиновое время и активированное частичное тромбопластиновое время (АЧТВ) не определялось из-за отсутствования свертывания крови, уровень фибриногена не определялся — не формировалось сгустка). Исследование миелограммы не проводилось из-за опасности провокации нового источника кровотечения. Потребовались перевод на искусственую внтиляцию легких, назначение Викасола, Апротекса, трансфузии свежезамороженной плазмы, препарата НовоСэвэн, эритроцитарной массы после индивидуального подбора 28 и 29.08.12 г.). Кроме того, с момента поступления осуществлялась антибактериальная терапия Меронемом, противовирусная — Зовираксом, преднизолоном 45 мг в сутки (2 мг/кг/сут) внутримышечно с постепенной отменой; флуконазолом, Эссенциале H.
В динамике состояние прогрессивно ухудшалось: появилась диффузная кровоточивость со слизистых рото- и носоглотки, проведенная тампонада не дала значимого эффекта. Стабилизировать состояние ребенка не удалось. Нарастал синдром полиорганной недостаточности: энцефалопатия вплоть до комы, интестинальная, сердечно-сосудистая, дыхательная, печеночная недостаточность с гипокоагуляцией, развитием ДВС-синдрома. Параклинически сохранялась глубокая гипокоагуляция, синдром цитолиза, гипопротеинемия, панцитопения. 30.08.12 в 23.00 развилось профузное легочное кровотечение. 31.08.12 в 2.15 наступила смерть при явлениях сердечно-сосудистой недостаточности.
Диагноз клинический. Основное заболевание: генерализованная вирусно-бактериальная инфекция тяжелой степени.
Осложнения: синдром полиорганной недостаточности (печеночная недостаточность, энцефалопатия, гастроинтестинальная, сердечно-сосудистая, дыхательная недостаточность, ДВС-синдром, панцитопения). ДВС-синдром.
При патологоанатомическом исследовании обнаружено:
Макроскопически: кожа бледная, с желтушным оттенком, мягкие ткани пастозны. В серозных полостях умеренное количество прозрачной желтоватой жидкости. Серозные оболочки блестящие, несколько иктеричны. В просвете трахеи, бронхов жидкая кровь. Легкие темно-красного цвета, тестоватой консистенции. Сердце конусовидной формы. Миокард серовато-розового цвета. Клапаны сформированы правильно, створки тонкие, полупрозрачные. В полостях небольшое количество жидкой крови. Селезенка — 70,0 г (норма 40,0 г), эластической консистенции, синюшно-вишневого цвета, на разрезе без соскоба пульпы. Лимфоузлы брыжейки, бифуркационные паратрахеальные увеличены до 0,8 см, серовато-розовые. Вилочковая железа серовато-белая, 20,0 г. Слизистая пищевода с продольной складчатостью, с рассеянными эрозиями во всех отделах. В желудке кровь и свежие сгустки, слизистая с крупными эрозиями. Слизистая кишечника гиперемирована, складчатая, кишечное содержимое с примесью крови. Печень увеличена, 600 г (норма 500), с закругленным передним краем, эластической консистенции, желтовато-коричневого цвета, с гладкой поверхностью, на разрезе полнокровна. Желчевыводящие пути проходимы. Поджелудочная железа дольчатая, серовато-белая. Почки бобовидной формы, эластической консистенции, красного цвета. На разрезе малокровны, рисунок сохранен. Надпочечники листовидной формы, корковый слой желтого цвета. Полость черепа не вскрывалась.
Кишечник. В тонкой кишке в собственном слизистом слое полнокровие, мелкие кровоизлияния, рассеянная лимфомононуклеарная инфильтрация с примесью плазматических клеток. Дистрофические изменения клеток эпителия со слущиванием их на вершинах ворсинок. В солитарных фолликулах гиперплазия лимфоидной ткани, очаговые мононуклеарные скопления. В толстой кишке слизистая сохраняет строение, мелкие кровоизлияния в собственном слое слизистой.
Печень. Тотальная средне- и крупнокапельная жировая дистрофия гепатоцитов. В дольках очаговые лимфомононуклеарные скопления, фокальные некрозы гепатоцитов, много крупных клеток ретикуло-эндотелиальной системы, некоторые с явлениями эритрофагии. Отмечается рассеянная инфильтрация лимфоцитами и мононуклеарами портальных трактов.
Костный мозг. Клеточный состав представлен всеми ростками с умеренной гиперплазией и омоложением. Выявляются очаговые некрозы кроветворной ткани.
Лимфоузлы. Рисунок сохранен. Единичные фолликулы со светлыми центрами. В кортикальном и паракортикальном слоях микроабсцессы. В синусах множество макрофагов с наличием гемофагоцитоза (рис. 1).
.jpg)
Селезенка. Лимфоидные фолликулы без светлых центров. Отмечается пролиферация ретикулярных клеток, значительная инфильтрация мононуклеарными клетками с активным гемофагоцитозом, множественные микроабсцессы.
Вилочковая железа. Акцидентальная инволюция 4–5 степени: дольки уменьшены, разделены широкими тяжами фиброзной стромы, тельца Гассаля расширены. В клеточном составе большое количество крупных мононуклеарных фагоцитов с признаками эритрофагии (рис. 2).
.jpg)
Легкие. Преобладают альвеолы, заполненные кровью. Встречаются очаговые параваскулярные лимфомононуклеарные скопления.
Желудок. В слизистой острые эрозии, очаговые кровоизлияния.
Почки. Структура сохранена, клубочки малокровные, дистрофия эпителия канальцев.
Миокард. Выраженная дистрофия кардиомиоцитов. В миокарде интерстициальная реакция в виде мелких скоплений лимфомононуклеаров.
Из ткани печени, селезенки, легкого выделена Salmonella enteritidis.
Патологоанатомический диагноз: сальмонеллез (Salmonella enteritidis), септическая форма, гиперплазия лимфоидной ткани кишечника, лимфоузлов, селезенки, легких, микроабсцессы в брыжеечных лимфоузлах, селезенке, очаговые некрозы в костном мозге. Фоновое заболевание: иммунодефицитное состояние, пролиферация клеточных компонентов системы мононуклеарных фагоцитов с явлениями эритрофагии в лимфоузлах, вилочковой железе, печени.
Осложнения: ДВС-синдром: кровоизлияния в легкие, слизистую бронхов, пищевода, желудка, кишечника, острые эрозии слизистой пищевода, желудка. Выраженные дистрофические изменения внутренних органов. Респираторный дистресс-синдром: гиалиновые мембраны в легких.
Резюме. На основании данных анамнеза, клинического течения болезни, результатов клинико-лабораторного и патологоанатомического обследования был установлен диагноз сальмонеллеза, септикопиемического варианта с высевом из органов Salmonellaе enteritidis, осложненного гемофагоцитарным синдромом.
Диагноз подтвержден на аутопсии обнаружением в пораженных органах (тимус, селезенка, лимфатические узлы) массивной лимфомакрофагальной инфильтрации с явлениями гемофагоцитоза и очаговыми некрозами в костном мозге.
Имелись объективные трудности диагностики сальмонеллезной инфекции, в частности, отсутствия ее манифестации диарейным синдромом, исключительной редкостью септикопиемического варианта течения сальмонеллеза, вызванного Salmonellae enteritidis.
Генерализованная форма сальмонеллеза обусловлена дефектами в системе иммунитета, при ней формируется длительная бактериемия (которая бывает всегда кратковременной при локализованных гастроинтестинальных формах — гастроэнтерите и гастроэнтероколите). Интоксикационный синдром определяет эндотоксинемия, обусловленная разрушением энтеро- и цитотоксинов, продуцирующихся живыми сальмонеллами. В настоящее время отмечается особо, что септические формы сальмонеллеза характерны для ВИЧ-инфекции в стадии СПИДа. Ввиду этого, больной с сальмонеллезным сепсисом всегда должен обследоваться на ВИЧ-инфекцию. Такие инфекции, как малярия, гистоплазмоз, серповидно-клеточная анемия и другие, характеризующиеся перегрузкой фагоцитов, также предрасполагают к инвазивному сальмонеллезу.
В данном случае, несомненно, следовало провести дифференциальный диагноз между вторичным и первичным ГФС, так как при обеих нозологиях пусковым механизмом заболевания может быть инфекция, а клиническое течение, лабораторные данные, морфологическая характеристика часто схожи. Основным отличием двух форм ГФС может быть персистирующий дефицит NK-клеток, который наблюдается при первичном и не выражен при вторичном ГФС. Поскольку иммунологическое обследование не проводилось, исключить наследственный характер заболевания не представлялось возможным. Сложность ведения больных с ГФС в отделениях реанимации определяется следующими положениями:
- Критерии диагностики не являются высокоспецифичными (клиника сепсиса, СПОН, синдром системного воспалительного ответа (англ. systemic inflammatory responce syndrome — SIRS)). Нет четкой клинической границы между этими синдромами.
- Сепсис — более частый диагноз, не предполагающий назначения необходимой для лечения ГФС иммуносупрессивной терапии (дексаметазон, циклоспорин, этопозид, трансплантация кроветворных стволовых клеток).
- Принципиальной позицией является положение о безуспешности посиндромной и органозамещающей терапии без назначения иммуносупрессивной терапии.
- Тяжелая комбинированная патология гемостаза осложняет проведение любых инвазивных манипуляций и требует агрессивной трансфузионной поддержки на основании точного мониторинга гемостаза в реальном времени [2].
Литература
- Arceci R. J. When T cells and macrophages do not talk: the hemophagocytic syndromes // Curr Opin Hematol. 2008; 15 (4): 359–367.
- Мосчан М. А., Полтавец Н. В. ГФС в неотложной и интенсивной терапии // Педиатрическая фармакология. 2011; 8 (2); 85–88.
- Охотникова Е. Н. и соавт. Гемофагоцитарный синдром в педиатрической практике // Педиатрия. 2011; 90 (4); 61–70.
- Малышенкова Н. К. Клинические формы хронической Эпштейна–Барр вирусной инфекции: вопросы диагностики и лечения // Лечащий Врач. 2003. № 9. С. 32–38.
- Анохин В. А., Фаткуллина Г. Р., Акчурина Л. Б. Гемофагоцитарный синдром и герпес-вирусные инфекции // Журнал инфектологии. 2012; 4 (1); 81–83.
Е. И. Краснова 1 , доктор медицинских наук, профессор
Т. И. Белоусова
С. А. Лоскутова, доктор медицинских наук, профессор
В. Г. Кузнецова, доктор медицинских наук, профессор
ГБОУ ВПО НГМУ МЗ РФ, Новосибирск
ГФС (синоним гемофагоцитарный лимфогистиоцитоз) является нарушением иммунной регуляции, вызванным провоспалительными цитокинами, при сопутствующей патологии иммунных клеток (NK и цитотоксических лимфоцитов Т), обусловленной генетически (врождённая форма, наблюдается главным образом у детей, в более лёгких формах также у взрослых) или приобретённой (поражение этих клеток, вызванное тяжёлой инфекцией [чаще всего ВЭБ], аутоиммунным заболеванием или злокачественным новообразованием [особенно лимфомой]). Редко субстратом ГФС являются врожденные иммунодефициты. Идиопатические случаи составляют 6–18 %. Выделяемые в механизме замкнутого круга во всё большем и большем количестве провоспалительные цитокины вызывают патологически усиленную общесистемную воспалительную реакцию, что ведёт к повреждению всех органов.
Разновидностью ГФС является синдром активации макрофагов (САМ) →разд. 16.3, который отличается от типичного ГФС высокой концентрацией СРБ в плазме. Наблюдается чаще всего в течении болезни Стилла, системной красной волчанки, а также после пересадки костного мозга.
КЛИНИЧЕСКАЯ КАРТИНА И ЕСТЕСТВЕННОЕ ТЕЧЕНИЕ
Постоянная лихорадка, увеличение печени и селезёнки; можно также обнаружить симптомы геморрагического диатеза (не всегда), бледность и/или желтушность кожных покровов, иногда отёки, эритематозная, папулёзная или уртикарная сыпь (бывает кровоточащая), экссудаты в полостях тела и нарушения сознания. Без лечения летальность составляет 100 %. наверх
Дополнительные методы исследования
1. Лабораторные исследования:
1) общий анализ периферической крови с мазком — панцитопения (в т. ч. обычно лимфопения);
2) биохимические исследования →Диагностические критерии;
3) могут появляться нарушения гемостаза, соответствующие синдрому ДВС →разд. 15.21.
2. Морфологические исследования: в костном мозге, селезёнке, лимфоузлах, иногда в других органах или в спинномозговой жидкости (СМЖ; в случае поражения ЦНС) можно обнаружить гемофагоцитоз макрофагов, в цитоплазме которых видны поглощённые эритроциты, иногда также другие клетки (напр. лейкоциты, тромбоциты) или их фрагменты.
3. Другие исследования: с целью проведения дифференциальной диагностики →см. ниже.
Молекулярный диагноз (обнаружение соответствующей мутации) или соответствие ≥5 из 8 критериев:
1) лихорадка ≥38,5 °C;
2) увеличение селезёнки;
3) цитопения в периферической крови ≥2 из 3 линий гемопоэза (гемоглобин 2000 нг/мл, для постановки диагноза достаточным является наличие 4 критериев.
Калькулятор, вычисляющий вероятность диагноза, приобретенного HLH на основе клинических данных отдельного пациента (HScore) →http://saintantoine.aphp.fr/score/.
С целью дифференциации причин отдельных отклонений, составляющих диагностические критерии, следует провести:
1) лабораторные исследования — число ретикулоцитов, СОЭ, биохимические исследования (ЛДГ, аминотрансферазы, билирубин, креатинин, мочевина, СРБ), исследования системы свёртывания, электрофорез белков сыворотки, иммуноглобулины, реакция Кумбса;
2) вирусологические исследования, в т. ч. на ВЭБ, в том числе иммуногистохимическое исследование тканевого материала на наличие LMP1;
3) неврологическое обследование, в случае симптомов со стороны ЦНС исследование СМЖ с оценкой мазка;
4) УЗИ и/или КТ селезёнки и печени, возможно КТ или МРТ головы;
5) аспирационная биопсия и трепанобиопсия костного мозга;
6) другие исследования, исходя из непосредственно определяемых показаний, в т. ч. поиски лимфомы (гистологическое исследование увеличенного лимфоузла).
Самым важным состоянием для дифференциальной диагностики с HLH (также может сопутствовать) является сепсис →разд. 18.8.
Другие причины повышенного уровня ферритина (→разд. 27.1), включая болезнь Стилла, которая часто вызывает HLH.
Проводится в гематологическом центре. наверх
1. Этиотропное лечение: протокол HLH-2004 (этопозид, дексаметазон, циклоспорин); в течение 8 нед.; в случае поражения ЦНС → метотрексат интратекально. В случае резистентности к этому лечению можно применить протоколы химиотерапии, используемые при лечении лимфом, и алемтузумаб, а также моноклональные антитела анти-IL-6, в последующем алло-ТГСК. В случае больных, у которых ГФС развился на фоне лимфомы или другого новообразования, вместо протокола HLH-2004 примените стандартный протокол лечения данного новообразования. Если ГФС является вторичным к инфекции ВЭБ → рассмотрите применение ритуксимаба. В случае рецидива → повторите вышеуказанную схему, в последующем — алло-ТГСК. Лечение САМ →разд. 16.3.
2. Симптоматическое лечение: плазмаферез, переливание ВВИГ и компонентов крови, изоляция в абактериальных условиях, применение противобактериальных, противогрибковых и противовирусных лекарств, жаропонижающие лекарства.
Гемофагоцитарный лимфогистиоцитоз ( HLH ), также известный как haemophagocytic лимфогистиоцитоз ( британское правописание ), и гемофагоцитарная или haemophagocytic синдрома , является редким расстройством гематологического видело чаще у детей , чем у взрослых. Это угрожающее жизни заболевание тяжелой гипервоспаления , вызванной неконтролируемой пролиферации активированных лимфоцитов и макрофагов , характеризующихся пролиферацией морфологически доброкачественных лимфоцитов и макрофагов , которые секретируют большие количества воспалительных цитокинов. Он классифицируется как один из гиперцитокинемии синдромов.
содержание
классификация
Первичный HLH, также известный как семейная haemophagocytic лимфогистиоцитоз (FHL) или семейная erythrophagocytic лимфогистиоцитоз, представляет собой гетерогенное аутосомно - рецессивное заболевание обнаружено, что более распространенным с родительской единокровностью.
Вторичный haemophagocytic лимфогистиоцитоз (приобретенный haemophagocytic лимфогистиоцитоза) происходит после сильной иммунологической активации, таких как то, что может произойти с системной инфекцией, иммунодефицитом, или основной злокачественной опухолью.
Обе формы характеризуются подавляющим активацией нормального Т-лимфоцитов и макрофагов, неизменно приводят к клиническим и гематологическим изменениям и смерти при отсутствии лечения.
Первичная HLH вызвана потерей функции (т.е. инактивировать) мутацию в генах, цитотоксические Т и / или NK - клетка использует , чтобы убить целевые клетки , такие как те , которые заражены возбудителями , например, вирус Эпштейна-Барр (EBV) или вирус денге . Эти мутации включают в себя те , в следующих генов: UNC13D , STX11 , RAB27A , STXBP2 , Lyst , PRF1 1, SH2D1A , BIRC4 , ITK , CD27 , и MAGT1 . Вторичный HLH связан с и как полагают, способствует злокачественных и доброкачественных заболеваний , которые аналогичным образом ослабляют иммунную систему способность «ы , чтобы атаковать EBV-инфицированных клеток. Злокачественные расстройства , связанные с вторичной чЛГ включают Т - клеточную лимфому , лимфому B , острый лимфобластный лейкоз , острый миелобластный лейкоз , а также миелодиспластический синдром . Незлокачественные расстройства , связанные со вторичной чЛГ включают: аутоиммунные нарушения , такие как ювенильный идиопатический артрит , ювенильные болезни Кавасаки , системную красную волчанку , в юношеских и взрослой протекающих формах болезнь Стилла , и ревматоидный артрит ; иммунодефицитные расстройства , такие как тяжелый комбинированный иммунодефицит , синдром DiGeorge , синдром Вискотта-Олдрича , атаксия телеангиэктазии , и дискератоз Врожденный ); и инфекций , вызванных EBV, цитомегаловирус , ВИЧ / СПИД , бактерий , простейших и грибов . Вторичный HLH может также быть результатом ятрогенных причин , таких как костный мозг или другие трансплантации органов; химиотерапия; или терапия с иммунодепрессивными агентами; Около 33% всех случаев чЛГа,
75% азиатских случаев чЛГа, и почти 100% случаев чЛГа вызванных мутаций в SH2D1A (см X-связанный лимфопролиферативной болезнь типа 1 ) связаны с, и мыслью вызвали или способствовал, EBV инфекционное заболевание. Эти случаи чЛГ классифицируются как принадлежащие к классу Эпштейна-Барр вирус-ассоциированных лимфопролиферативных заболеваний и называется EBV + чЛГ .
Признаки и симптомы
Наступление чЛГ происходит в возрасте до 1 года в
70% случаев. Семейная HLH следует заподозрить, если братья и сестра с диагнозом чЛГом или если симптомы повторились, когда терапия была прекращены. Каждый полный брат ребенка с семейным чЛГом имеет 25% вероятность развития заболевания, 50% шанс нести дефектный ген (который очень редко, связанный с каким-либо риском заболевания) и 25% шансом не затронули а и не несущий дефект гена.
Пациенты с чЛГ, особенно при отсутствии лечения, может потребоваться интенсивная терапия. Таким образом, HLH должны быть включены в дифференциальной диагностике интенсивной терапии (отделение интенсивной терапии) пациентов с цитопении и hyperferritinemia.
генетика
Пяти генетические подтипы (FHL1, FHL2, FHL3, FHL4 и FHL5) описаны, с предполагаемой распространенностью одного в 50000 и равным распределением полов. Молекулярно - генетическое тестирование для четырех причинных генов, PRF1 (FHL2), UNC13D (FHL3), STX11 (FHL4) и STXBP2 (FHL5), доступен на клинической базе. Симптомы FHL обычно проявляется в течение первых нескольких месяцев жизни и даже могут развиваться в утробе матери . Тем не менее, симптоматическая презентации в детстве и даже в молодом возрасте наблюдается в некоторых случаях.
Пять подтипов FHL каждый связаны с конкретным геном:
Почти половина случаев 2 типа семейного гемофагоцитарного лимфогистиоцитоза обусловлены би-аллельные PRF1 мутаций.
диагностика
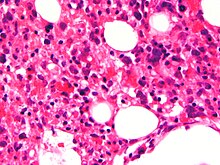
Анализ крови , как правило , показывает , уменьшился количество клеток крови -включая пониженное количество циркулирующего красные кровяные клетки , белые клетки крови и тромбоциты .
Костный мозг может показать гемофагоцитоз .
Функциональные пробы печени обычно повышены. Низкий уровень белка альбумина в крови является общим.
Сывороточного С - реактивный белок , СОЭ и ферритина уровня заметно повышен. У детей ферритина выше 10000 является очень чувствительной и специфичной для диагностики чЛГа, однако, диагностическая утилита для ферритина менее для пациентов чЛГа взрослых.
В сыворотке крови фибриногена уровень обычно низок , а D-димер уровень повышен.
Биопсии костного мозга показывает гистиоцитоз .
В настоящее время (2008) диагностические критериев для чЛГа являются
1. Молекулярная диагностика в соответствии с чЛГом. Они включают в себя идентификацию патологических мутаций PRF1, UNC13D или STX11.
2. Совершение пяти из восьми критериев ниже:
- Лихорадка (определяется как температура> 100,4 ° F,> 38 ° С)
- Увеличение селезенки
- Снижение количества клеток крови влияет, по крайней мере два из трех линий в периферической крови:
- Гемоглобин 9 / л ( тромбоцитопения )
- Нейтрофилы 9 / л ( нейтропения )
- Высокие уровни в крови триглицеридов (натощак, превышающие или равные 265 мг / 100 мл) и / или снижение количества фибриногена в крови (≤ 150 мг / 100 мл)
- Ферритин ≥ 500 нг / мл
- Haemophagocytosis в костном мозге , селезенка или лимфатических узлах
- Низкая или отсутствуют естественные киллеры активность
- Растворимые CD25 (растворимый рецептор IL-2)> 2400 Ед / мл (или на местной опорной лаборатории)
Кроме того, в случае семейного чЛГа, никаких признаков злокачественной опухоли не должны быть очевидны.
Не все пяти из восьми критериев, необходимые для диагностики чЛГа у взрослых, и высокий индекс подозрений для постановки диагноза требуется , как задержка приводит к увеличению смертности. Диагностические критерии были разработаны в педиатрической популяции и не были утверждены для взрослых пациентов чЛГа. Попытки улучшить диагностику чЛГ включали использование HScore , которые могут быть использованы для оценки риска индивида в чЛГ. У взрослых, растворимый IL-2 , было установлено , чтобы быть очень чувствительным маркером чЛГ, демонстрируя чувствительность 100% ниже отсечки 2400 ед / мл.
Дифференциальная диагностика чЛГа включает в себя вторичный чЛГ и синдром макрофагальных активаций или другие первичные иммунодефициты , которые представляют с гемофагоцитарным лимфогистиоцитозом, такие как Х-хромосомой болезнь лимфопролиферативной .
Другие условия , которые могут быть перепутаны с этим условием включают синдром аутоиммунного лимфопролиферативных . Как синдром интенсивного воспаления его необходимо дифференцировать от сепсиса , что может быть чрезвычайно сложной задачей.
Диагноз приобретенного, или вторичного, чЛГ, как правило, в сочетании с инфекцией вирусами, бактериями, грибами, или паразитов , или в сочетании с лимфомой, аутоиммунного заболевания, или болезни обмена веществ. Приобретенный HLH может быть снижен, нормальным, или увеличение клеток NK - активности.
Основной дифференциальный диагноз чЛГ является синдром Griscelli (тип 2). Это редкое аутосомно - рецессивное заболевание характеризуется частичным альбинизм, гепатоспленомегалия, панцитопения, гепатит, иммунологическими аномалиями, и лимфогистиоцитозом. В большинстве случаев были диагностированы от 4 месяцев до 7 лет, со средним возрастом около 17 месяцев.
Три типа синдрома Griscelli признается: тип 1 имеет неврологическую симптоматику и мутацию в MYO5A . Прогноз зависит от тяжести неврологических проявлений. Тип 2 имеет мутации в RAB27A и haemophagocytic синдрома, с аномальными Т-клетками и активацией макрофагов. Этот тип имеет серьезный прогноз , если не лечить. Тип 3 имеет мутации в melanophilin и характеризуется частичной альбинизм. Этот тип не представляет угрозу для тех , кто так подействовало.
лечение
В средних случаях лечение от причины, где это возможно, указывается. Кроме того, лечение самого чЛГ обычно требуется.
В то время как оптимальное лечение чЛГ все еще обсуждается, текущие режимы лечения обычно включают высокую дозу кортикостероидов , этопозид и циклоспорин . Внутривенный иммуноглобулин также используется. Метотрексат и винкристин также были использованы. Другие лекарства включают цитокина целевой терапии .
С 20 ноября 2018 г. FDA одобрил моноклональные антитела анти-IFN-гамма emapalumab (фирменное наименование Gamifant) для лечения детей и взрослых первичного чЛГа.
Прогноз
Прогноз охраняется с общей смертности на 50%. Плохие прогностические факторы включают чЛГ, связанный со злокачественной опухолью, с половиной пациенты умирают в 1,4 месяцев по сравнению с 22,8 месяцев, не ассоциированные с опухолью пациентов чЛГа.
Вторичный HLH у некоторых людей может быть самоограниченным , потому что пациенты имеют возможность полностью восстановиться после получения только поддерживающего лечения (т.е. IV иммуноглобулина только). Однако длительные ремиссии без применения цитотоксических и иммуно-супрессивной терапии вряд ли в большинстве взрослых с чЛГ и в тех , с вовлечением центральной нервной системы (головного и / или спинного мозга).
история
Первый случай доклад чЛГ был опубликован в 1952 году.
Смотрите также
Исследование
Систематический обзор недавно сообщил, объединенные доли являются высокой температурой 97,2%, 70,2% гепатомегалия, спленомегалия 78,4%, тромбоцитопения 90,1%, анемия 76,0%, и ферритина в сыворотке ≥500 мкг / л 97,1%. Летальность составляет 14,6% среди пациентов денге гемофагоцитарного лимфогистиоцитоза.
Читайте также:
