Филогенетический анализ вирусов для
Основным инструментом филогенетического анализа является сравнение близких по структуре или по функции генов или белков, и прежде всего, сравнение их первичных последовательностей. Важнейшим свойством функционально значимых структур макромолекул является их эволюционный консерватизм. Чем меньше функциональная важность отдельных участков генов, тем больше они имеют тенденцию к эволюционной изменчивости. Так, например, псевдогены по-видимому полностью утратили функциональную активность. Для них характерно быстрое накопление в ходе эволюции различных замен, делеций и вставок, разрушающих исходную структуру гена. С другой стороны, гистоны Н4, играющие важную роль в упаковке хроматина, почти не изменялись на протяжении всей эволюции животных. Консервативность генов позволяет выявить отдаленное родство между их представителями, давно разошедшимися в ходе эволюции и выполняющими иногда разные функции. Однако для филогенетического анализа необходимо и наличие определенного уровня изменчивости генов. Мутации, делеции и вставки являются своего рода метками, благодаря которым удается восстановить пути эволюции современных форм макромолекул. Гены с разной величиной консервативности пригодны для изучения разных эволюционных уровней. Сильно консервативные гены и их продукты (гистоны, тРНК) нельзя , например, использовать для исследования эволюции отрядов и более мелких таксонов, но с успехом можно применять для изучения эволюции более крупных таксонов. Сильно вариабельные гены, наоборот, дают хорошее разрешение лишь на поздних эволюционных этапах. В последнее время метод полимеразной цепной реакции (ПЦР) с последующим анализом нуклеотидной последовательности широко используется для точной идентификации вирусов и определения их родства в отношении других штаммов. Для сравнительной характеристики геномов различных штаммов вирусов также проводят рестрикционный анализ ПЦР-продуктов. Обычно для построения филогенетического дерева используются данные последовательностей нуклеиновых кислот. Филогенетическое дерево очень ясно показывает родство между вирусами, если анализируется большое количество изолятов. Для этих целей существует много компьютерных программ. Наиболее популярные пакеты программ- PHYLIP(PHYLogeny Inference Package), PAUP(Phylogentic Analysis Using Parsimong),CLUSTAL и MEGA. Для филогенетического анализа особенно интересны РНК-содержащие вирусы, которые существуют как гетерогенные популяции. Их геном более генетически пластичен, чем геном ДНК-содержащих вирусов. Так, геном вируса бешенства, который относится к роду Lyssavirus семейства Rhabdoviridae, представлен одноцепочечной негативной РНК длиной около 12000 пар оснований (п.о) , кодирующей пять основных белков. Белки подразделяются на три функциональные группы: оболочечные (G ,M) нуклеокапсидный (N) и РНК-полимеразный комплекс, состоящий из L и NS белков. Белки N,NS и L вместе с вирионной РНК образуют нуклеокапсид , который окружен мембраной, содержащей трансмембранный гликопротеин G , ответственный за антигенные свойства вируса. Существование псевдогена ( между G и L цистронами , является отличительной особенностью вируса бешенства от вируса везикулярного стоматита. N-ген лиссавирусов, как показало клонирование и секвенирование, является наиболее консервативным по своей структуре из всех генов вируса бешенства. Mannen K.et al, в 1991 г. определили большую зависимость различий в N-гене от географической локализации, чем от хозяйской специфичности. В Онтарио вирус бешенства, который описан как единственный "Арктический" тип, разделили на четыре основных типа. Эти типы филогенетически разветвляются на две основные ветви , одна из которых состовляет один тип, вторая три основных типа, что отражает историческое передвижение вируса в регионе от середины к концу 50-х гг. Эпизоотия передвигалась на юг Онтарио с севера и Квебека. Изменения в последовательности N-гена , определенных для 4-х вирусных типов, могут представлять генетический маркер для более существенных изменений в других частях вирусного генома. Kissi et al представили первое сравнение между генотипами и молекулярными различиями N-гена внутри первого генотипа. Филогенетический анализ гена нуклеопротеина 82-х лиссавирусов подтвердил существование шести генотипов лиссавирусов, и также выделил изоляты бешенства первого генотипа в отдельные генетические линии. Изоляты с меньшей чем 80% нуклеотидной и 92% аминокислотной гомологией относятся к различным генотипам. Два коротких региона из 400 нуклеотидов, кодирующих аминоконец N-протеина, и 93 нуклеотида, кодирующих N-NS-регион, могут быть использованы для определения географического распределения основных вирусных линий. Выявлено два региона, имеющих наименьший уровень гомологии: участок длиной 199 пар оснований (нуклеотиды от 1080-го до 1278-го) и более протяженный фрагмент, расположенный между 99-м и 405-м нуклеотидами. Филогенетическое дерево построили, используя пакет программ MEGA. С их помощью получили дерево с ветвями, делящимися на 6 кластеров, которые соответствуют 6-ти генотипам. Сравнение изолятов показало, что в генетическом плане наиболее близкими оказались 4-й и 5-й генотипы, у которых уровень различий составил 79,8% (нуклеотидный уровень) и 93,3% (аминокислотный уровень) [Duvenhage virus (4) и EBL1(5)]. Внутри генотипа 1 наименьшее сходство среди изолятов из Азии и Латинской Америки - 83,3%; и наибольшее сходство в изолятах из Африки и Латинской Америки - 92,2%. Филогенетический анализ изолятов 1-го генотипа вируса бешенства. Kissi et al. идентифицировали 11 филогенетических линий, взятых в соответствии с их географическим происхождением и видом хозяина: Африка 1а, Африка 1в, Африка 2, Африка 3, Азия, Арктика, Европа/Средний Восток, Латинская Америка 1 и 2 и две группы вакцинных штаммов. Филогенетическое дерево строилось на основании сравнения фрагмента или целого N-гена. Этот анализ позволил более точно определить циркуляцию по зонам и установить происхождение и распространение бешенства для некоторых линий, которые, возможно, произошли независимо на этом континенте от различных предшественников. Хотя циркуляция африканских вирусов 1а и 1в более отлично от вирусов, распространенных в Европе и Среднем Востоке, однако была выявлена генетическая связь между ними, что свидетельствует об общем предке. Выяснено, что накопление большинства нейтральных мутаций в географически разделенных вирусных популяциях привело к значительным расхождениям в нуклеотидной последовательности гена нуклеопротеина. При исследовании гена нуклеопротеина 11-ти вирусов бешенства японскими учеными было выявлено 9 отдельных кластеров по гомологии менее 90% региона N-гена. Тем самым они подтвердили данные филогенетического анализа, полученные Kissi et al. Таким образом, варианты вируса бешенства, сгруппированные в соответствии с их географическим распределением, могут быть использованы для исследования эволюционного развития вируса бешенства.
Принципы и методы ОТ-ПЦР.
Обратно-транскриптазная ПЦР состоит из двух этапов:
-
1- синтез комплементарной ДНК с помощью фермента обратной транскриптазы и затравки
2- амплификация гена или его фрагментов при помощи фермента термостабильной ДНК-полимеразы и коротких олигонуклеотидных 20-30-членных затравок (праймеров), комплементарных 3'- концевым последовательностям антипаралельных цепей ДНК гена. Повторяя стадии денатурации, отжига праймеров и полимеризации (достройка праймеров) 30-35 раз за 2-3 часа получают миллионы копий специфического участка генома вирусов и бактерий.
Аликвоты ПЦР-продуктов разрезают соответствующими ферментами и разделяют в 1%-м агарозном геле. Учет результатов проводят по размеру ПЦР-продуктов с помощью электрофореза в агарозном геле. На основании размеров и расстояний пробегов маркерных ДНК вычисляют размеры исследуемых фрагментов ДНК.
Анализ источника вируса иммунодефицита человека
В этом примере показано, как создать филогенетические деревья из нескольких деформаций HIV и вирусов SIV.
Мутации накапливаются в геномах патогенов, в этом случае человеческого/обезьяноподобного вируса иммунодефицита, во время распространения заражения. Эта информация может использоваться, чтобы изучить историю событий передачи, и также как доказательство для источников различных вирусных деформаций.
Существует две охарактеризованных деформации человеческих вирусов AIDS: тип 1 (HIV-1) и тип 2 (HIV-2). Обе деформации представляют заражения перекрестных разновидностей. Водохранилище примата HIV-2 было ясно идентифицировано как закопченный mangabey (Cercocebus atys). Источник HIV-1, как полагают, является обыкновенным шимпанзе (Троглодиты панорамирования).
В этом примере изменения трех самых длинных областей кодирования от семнадцати различных изолированных деформаций Человеческого и Обезьяноподобного вируса иммунодефицита используются, чтобы создать филогенетическое дерево. Последовательности для этих штаммов вируса могут быть получены из GenBank® с помощью своих инвентарных номеров. Три необходимых области кодирования, белок затычки, политический полибелок и предшественник полибелка конверта, могут затем быть извлечены из последовательностей с помощью информации о CDS в записях GenBank.
Можно использовать getgenbank функционируйте, чтобы скопировать данные GenBank в структуру в MATLAB®. Поле SearchURL структуры содержит адрес фактической записи GenBank. Можно просмотреть эту запись с помощью web команда.
Получите информацию о последовательности из базы данных NCBI GenBank для остальной части инвентарных номеров.
Для вашего удобства ранее загруженные последовательности включены в MAT-файл. Обратите внимание на то, что данные в общедоступных репозиториях часто курируются и обновляются; поэтому результаты этого примера могут немного отличаться, когда вы используете актуальные наборы данных.
Извлеките CDS для GAG, POL и областей кодирования ENV. Затем извлеките последовательности нуклеотида с помощью указателей CDS.
seqpdist и seqlinkage команды используются, чтобы создать филогенетическое дерево для GAG, кодирующего область с помощью метода 'Tajima-Nei', чтобы измерить расстояние между последовательностями и невзвешенным парным групповым методом с помощью средних арифметических или метода 'UPGMA', для иерархической кластеризации. Метод 'Tajima-Nei' только задан для нуклеотидов, поэтому последовательности нуклеотида используются, а не переведенные последовательности аминокислот. Расчет расстояния может занять довольно много минут, когда это очень в вычислительном отношении интенсивно.

Затем создайте филогенетическое дерево для полибелков POL с помощью метода 'Jukes-Cantor', чтобы измерить расстояние между последовательностями и взвешенным парным групповым методом с помощью средних арифметических или метода 'WPGMA', для иерархической кластеризации. Метод 'Jukes-Cantor' задан для последовательностей аминокислот, который, будучи значительно короче, чем соответствующие последовательности нуклеотида, означает, что расчет попарных расстояний будет значительно быстрее.
Преобразуйте последовательности нуклеотида в последовательности аминокислот с помощью nt2aa .
Вычислите расстояние и рычажное устройство, и затем сгенерируйте дерево.

Создайте филогенетическое дерево для полибелков ENV с помощью нормированных попарных баллов выравнивания в качестве расстояний между последовательностями и 'UPGMA', методом для иерархической кластеризации.

Эти три дерева подобны, но существуют некоторые интересные различия. Например, в дереве POL, последовательность 'сфинкса SIVmnd5440 Mandrillus ' помещается близко к деформациям HIV-1, но в дереве ENV это показывается как являющийся очень удаленным к HIV 1 последовательность. Учитывая, что эти три дерева показывают немного отличающиеся результаты, дерево согласия, использующее все три области, может дать лучшую общую информацию о полных вирусах. Дерево согласия может быть создано с помощью взвешенного среднего этих трех деревьев.
Обратите внимание на то, что различные метрики использовались в расчете попарных расстояний. Это могло сместить дерево согласия. Можно хотеть повторно вычислить расстояния для этих трех областей с помощью той же метрики, чтобы получить несмещенное дерево.

Филогенетическое дерево, следующее из нашего анализа, иллюстрирует присутствие двух кластеров и некоторых других изолированных деформаций. Самый компактный кластер включает все выборки HIV2; в верхней ветви этого кластера мы наблюдаем закопченный mangabey, который был идентифицирован как источник этого лентивируса в людях. Кластер, содержащий деформацию HIV1, однако не так компактен как кластер HIV2. От дерева кажется, что Шимпанзе является источником HIV1, однако, источник передачи перекрестных разновидностей людям является все еще вопросом дебатов среди исследователей HIV.

[1] Гао, F., и др., "Источник HIV-1 у шимпанзе троглодиты троглодитов Пэна", Природа, 397 (6718):436-41, 1999.
[2] Kestler, H.W., и др., "Сравнение обезьяноподобного вируса иммунодефицита изолирует", Природа, 331 (6157):619-22, 1998.
[3] Alizon, M., и др., "Генетическая изменчивость вируса AIDS: анализ последовательности нуклеотида два изолирует от африканских пациентов", Ячейка, 46 (1):63-74, 1986.
У вас есть модифицированная версия этого примера. Вы хотите открыть этот пример со своими редактированиями?
| Главная ≫ Инфотека ≫ Биология ≫ Происхождение жизни ≫ Молекулярная эволюция // Ратнер В. А. |
|
Теория эволюции является одним из краеугольных камней биологии и естествознания в целом. В последние годы теория эволюции испытывала ускоренное изменение и развитие в связи с появлением в естественнонаучной практике нового мощного пласта экспериментальных данных и теоретических методов. В поле зрения теории эволюции попали кодирующие макромолекулы: ДНК, РНК и белки, составляющие костяк организации клеток и организмов. Теперь уже широко известна роль ДНК и РНК как материальных носителей генов, как переносчика генетической информации от генов к белкам и белков как исполнителей самых разнообразных биологических функций: ферментов — катализаторов процессов обмена, антител — иммунных защитников организма, структурных элементов, регуляторных и транспортных Первые последовательности аминокислот в белках были расшифрованы нуклеотидов в генах и РНК — но массовое секвенирование началось только став к настоящему времени рутинной технологической операцией. Результаты секвенирования с тех пор накапливаются в компьютерных банках данных. К осени 1997 года крупнейшие международные банки данных содержали сведения примерно об одном миллиарде секвенированных нуклеотидов генов и других и о более Легко понять, что расшифровка нуклеотидных последовательностей генов , РНК и аминокислотных последовательностей белков создаёт новую ситуацию, позволяющую взглянуть на процесс эволюции с молекулярного уровня организации жизни. Действительно, в классической генетике аллели генов принято обозначать однородными буквами или индексами: B и b, Если в процессе эволюции один аллель вытесняет другого , то кажется, что происходят как бы утрата предыдущего и распространение . В то же время в молекулярной генетике известно, что многие мутации состоят в замене лишь одного или нескольких нуклеотидов: Поэтому замена аллеля может сводиться фактически к замене и фиксации единственного нуклеотида, а остальная, большая часть гена остаётся неизменной. Сходство последовательностей аллельных генов остаётся максимальным. Чем больше накапливается различий, тем меньше сходство последовательностей. Чем раньше дивергировали два гена, тем больше фиксированных различий они накопят. Таким образом, сходство последовательностей макромолекул можно положить в основу построений. Число различий мономеров к нему может быть мерой эволюционной дивергенции. Обилие секвенированных последовательностей в банках данных делает применимость этого подхода практически неограниченной. В течение последних 30 лет были разработаны несколько десятков компьютерных методов и пакетов программ для построения филогенетических деревьев макромолекул и филогенетического анализа, при помощи которых были построены многие сотни филогенетических деревьев макромолекул. В результате их анализа получены многочисленные интереснейшие данные, причём некоторые из них имели фундаментальное значение. Многие макромолекулы эволюционировали гораздо медленнее, чем морфологические признаки живых форм, поэтому их филогенетический анализ позволяет заглянуть в очень ранние периоды эволюционного процесса млн лет тому . Кодирующие макромолекулы эволюционируют с разными скоростями. Наиболее консервативными обычно являются гены и белки некоторых очень глубоких и рано возникших генетических процессов, которые представлены у многих форм жизни. Таковы, например, гены рибосомных РНК в молекулярный механизм синтеза , некоторых гистонов — белков компактизации ДНК хромосом. Менее консервативны гены и белки систем, которые встречаются у определённых широких групп видов , глобины . Наконец, наиболее изменчивыми являются гены и белки вирусов, которые стремительно изменяются в борьбе с иммунной системой их хозяев гриппа, , онкогенные . Этот важный результат сразу привлёк внимание к изучению архебактерий, причём вскоре было показано, что по многим другим свойствам они действительно существенно удалены как от эукариот, так и от эубактерий. Многие архебактерии существуют в природе в весьма экзотических условиях: при высокой температуре вблизи подводных вулканов; в среде, насыщенной метаном, соединениями Время разделения надцарств Bacteria и Archaea оценивается примерно в 3,5 млрд лет назад. Дерево Вууза включает в себя фракции кодируемых не только ядерными генами, но и генами клеточных органелл — митохондрий и хлоропластов. Легко заметить, что ядерная фракция кукурузы попадает в ветвь эукариот, как и ожидается, а фракции из митохондрий и хлоропластов этой же кукурузы mitochondrion, в ветвь эубактерий. Этот факт считается самым веским аргументом в пользу симбиотической гипотезы эволюционного возникновения эукариот, согласно которой митохондрии происходят от симбиотических предковых пурпурных бактерий, а хлоропласты — от цианобактерий , а вовсе не от ядерных структур эукариотических клеток. Белки-глобины (гемоглобины крови, миоглобины и их гены распространены в живой природе не столь широко. Они найдены в основном у животных, некоторые удалённые их представители найдены также у растений, а у прокариот они пока не обнаружены. Поэтому глобины образуют молекулярное надсемейство, охватывающее лишь часть таксономического дерева жизни. Деревья эволюции глобинов и их генов были построены многими исследователями. На рис. 2 приведено филогенетическое дерево глобинов, построенное А. Жарких в нашей лаборатории. В целом оно достаточно хорошо соответствует принятой таксономии животных. Из независимых палеонтологических данных можно почерпнуть датировки эпох существования общих предков многих ныне живущих биологических форм. Так, в дереве глобинов датированы шесть точек ветвления. Поскольку методы реконструкции деревьев позволяют определить длины рёбер дерева фиксированных замен, на различных маршрутах, то с учётом датировок это даёт возможность подсчитать скорости эволюции на разных этапах и в разных ветвях дерева. Нами получены очень любопытные . оказалось, что средняя скорость эволюции по всему белку была непостоянна и имела максимум примерно лет назад, в эпоху выхода позвоночных животных из Мирового океана на сушу . эта особенность выявилась ещё ярче для тех участков глобинов, которые отвечают за образование их четвертичной , структуры. Дело в том, что миоглобины и гемоглобины некоторых примитивных животных до сих пор представляют собой протомеры субъединицы . Круглоротые рыбы , имеют димерные агрегаты гемоглобинов, а большинство других позвоночных — тетрамерные агрегаты, состоящие из двух и двух . Субъединицы соединяются при помощи так называемых центров контакта, которые обозначаются Для современных гемоглобинов известны аминокислоты, входящие в центры контакта. Поэтому можно подсчитать скорости эволюции непосредственно для центров контакта. Максимум скорости чётко выявляется для центра контакта и регуляторного центра связывания дифосфоглицерата . Таким образом, при выходе позвоночных животных на сушу их гемоглобин приобрёл тетрамерную структуру, когда разные связаны между собой центрами и регуляторным центром ДФГ. Прежде всего в эту эпоху ген общего предка гемоглобинов был дуплицирован, а его копии в ходе дивергенции дали начало двум родственным . Именно в ту эпоху мы видим максимальную скорость эволюции центров их контакта и регуляции, а затем в течение сотен миллионов лет скорость резко падала, часто до нуля, когда возникшие центры были просто неизменны. Иначе говоря, мы как бы видим события эпохи формирования новых функциональных структур молекул гемоглобина, которые в дальнейшем сохраняются у всех наземных форм позвоночных. Следует учесть, что выход позвоночных на сушу и переход к дыханию свободным кислородом воздуха сопровождались резкой перестройкой всей системы дыхания, и в том числе структуры гемоглобинов. Ускорение эволюции в эту эпоху означает, что указанные приобретения были высоко адаптивными, то есть обеспечивали существенное преимущество их обладателям. Примером дерева наиболее быстрой молекулярной эволюции является дерево генов, кодирующих гемагглютинины H3 вируса , построенное Л.В. Омельянчуком и сотрудниками. Это белок вирусного капсида, некоторые выпуклые участки которого узнаются специфическими антителами хозяина . В результате иммунное сопротивление хозяина препятствует размножению вируса. Вирус гриппа имеет в котором мутации происходят с наибольшей известной частотой — на позицию, за репликацию. В XX веке изучены несколько локальных эпидемий и пандемий гриппа. Одной из первых изученных была пандемия испанки . В дальнейшем были зафиксированы пандемии гонконгского В большинстве случаев образцы эпидемических штаммов вируса гриппа были собраны и сохранены в коллекциях. После 1978 года РНК этих эпидемических штаммов были секвенированы, а позже теоретиками построены филогенетические деревья. Поскольку все эпидемии были датированы , оказалось возможным определить скорости эволюции. В эпидемических ветвях они оказались выше, чем в неэпидемических, причём особенно большое ускорение было выявлено именно в позициях, кодирующих антигенные детерминанты белка. Иммунное сопротивление хозяина размножению вируса является главным селективным фактором, действующим на вирус. Поэтому выплеск эпидемического штамма, не встречающего иммунного сопротивления хозяина, сопровождается ускорением эволюции. Это размножение высоко адаптивно для вируса и неадаптивно Напротив, после выработки иммунного ответа спокойное размножение вируса становится для него и хозяина нейтральным — в ожидании новых адаптивных мутаций и рекомбинаций. В целом можно говорить о коэволюции вируса и иммунного ответа системы хозяина. Вирус стремится как бы мутационно выскользнуть готового иммунного ответа хозяина, а хозяин стремится догнать новые варианты вируса путём выработки нового специфичного иммунного ответа. Ещё один пример — эволюция вируса ВИЧ иммунодефицита человека, HIV в английской , вызывающего заболевание СПИД приобретённого иммунодефицита, в английской транскрипции . Вирус ВИЧ передаётся половым путём, а также нередко при переливании крови, нестерильных инъекциях и хирургических вмешательствах, при родах от матери к ребёнку и в других подобных ситуациях. Он был обнаружен в начале в крови больных, погибавших от различных тяжёлых заболеваний. Эти заболевания имели одно общее свойство — иммунная система больного была неспособна противостоять как этому вирусу, так и сопровождавшим возбудителям других заболеваний, от которых, собственно, больные и погибали. Были обследованы образцы из банков переливания крови, которые созданы во многих странах для медицинских целей. Оказалось, что многие образцы содержали вирус ВИЧ. Различные штаммы вируса ВИЧ, выделенные в разных районах Африки, Карибского бассейна и США, были секвенированы, для них теми же методами, что и выше, построены филогенетические деревья из них показано . Это дерево указывает, что вирус ВИЧ существовал в Центральной Африке до 1960 года, был занесён на Гаити до середины и в США до 1978 года. Иначе говоря, его истоки лежат в Африке, где в некоторых странах вирусоносители составляют до половины населения, хотя заболевание СПИД проявляется далеко не у всех. Видимо, размножение вируса всё же сдерживается их иммунной системой. Вирус ВИЧ сходен с некоторыми вирусами обезьян, поэтому многие учёные предполагают, что он возник в результате изменчивости этих вирусов и был занесён в популяцию человека извне относительно недавно. Интересно, что изменчивость вируса ВИЧ столь велика, что он успевает измениться в сторону усиления своей агрессивности непосредственно в ходе развития болезни СПИД у некоторых больных в течение и менее. Оценки изменчивости показывают, что она превышает верхний допустимый предел, в результате чего иммунная система больного не успевает справиться с выработкой новых вариантов антител и фактически распадается, открывая путь процессу заболевания СПИД и других болезней. Именно в этом, состоит источник агрессивности заболевания СПИД. Есть биологические проблемы, которые в большой степени и всегда интересуют человека и человечество. Среди них особое место занимает проблема происхождения эволюции самого человечества. В последние годы в этой области прогресс связан в основном с филогенетическим анализом макромолекул. Американский учёный М. Гудмен и сотрудники построили филогенетические деревья для некоторых генов и белков от высших приматов и человека. На рис. 6 приведено одно из них. Из него следует, что порядок последовательного ответвления видов от эволюционного ствола человека таков: макак резус — орангутан — горилла — шимпанзе — человек. Иначе говоря, наиболее близким к человеку является шимпанзе. Следует отметить, что до этих работ в таксономии высших приматов выделяли два семейства: Hominidae, включавшее только вид Homo sapiens, и Pongidae, включавшее гориллу, шимпанзе и орангутана. анализ существенно изменил эту классификацию. Гудмен и его сотрудники предложили новую таксономию высших приматов:
Род Homo , Род Pan : обычный шимпанзе и карликовый ,
Близость человека и шимпанзе дала основание Дж. Дайамонду назвать человека третьим шимпанзе. По его оценкам, геномы человека и шимпанзе различаются примерно одиночных замен , то есть на каждый сотый нуклеотид, но остальные 99 из 100 нуклеотидов у них одинаковы. Время их молекулярной дивергенции оценивается лет назад. Эти факты являются очень веским аргументом в пользу естественного происхождения человека в процессе филогенетического развития жизни на Земле в противовес различным религиозным мифам о разовом творении тому назад. В заключение можно отметить, что молекулярный филогенетический анализ становится сейчас одним из весомых методов таксономии живых форм. Таксономия высших приматов сейчас больше базируется на молекулярных данных, чем других. В систематике наивысших таксонов, систематике высших растений решающую роль играют деревья рибосомных РНК. Таксономию бактерий вообще невозможно построить без молекулярных данных. Можно надеяться, что в дальнейшем роль этого подхода только возрастёт.
2.Айала Ф. Введение в популяционную и эволюционную генетику. . 3.Айала Ф., Кайгер Дж. Современная генетика. М.: Мир, 4.Кимура М. Молекулярная эволюция: Теория нейтральности. 5.Ратнер В.А., Жарких А.А., Колчанов И.А. и др. Проблемы теории молекулярной эволюции. Новосибирск: Наука, 1985. 6.Ратнер В.А. Краткий очерк теории молекулярной эволюции. Новосибирск: НГУ, 1992. 7.Ratner V.A., Zharkikh A.A., Kolchanov N.A. et al. Molecular Evolution. B. etc.: , 1996. Об авторе: Вирус болезни Ньюкасла (БН) широко распространенное заболевание, регистрируемое во многих странах и поражающее многие виды диких и домашних птиц. Несмотря на значительные успехи в диагностике, эпизоотологии, изучении патогенности вируса БН на молекулярном уровне, проблема борьбы с этим особо опасным заболеванием птиц остается актуальной [1, 2]. В основе профилактики болезни Ньюкасла лежат неспецифические и специфические средства защиты и методы их осуществления [3, 4]. Возбудитель БН – РНК-содержащий вирус (парамиксовирус птиц типа 1) являющийся таксоном рода Avulavirus, подсемейства Paramyxovirinae, семейства Paramyxoviridae, порядка Mononegavirales и характеризующийся минус-нитевым РНК-геномом. Его вирионная РНК представлена шестью генами, кодирующими гемагглютинин-нейраминидазу (HN), нуклеопротеин (NP), фосфопротеин (P), матриксный белок (М), РНК-зависимую РНК-полимеразу (L) и белок слияния (F), и двумя неструктурными белками V и W [5]. Для идентификации изолятов вируса болезни Ньюкасла были использованы полимеразная цепная реакция в реальном времени (РВПЦР) и антигенный анализ с помощью моноклональных антител. Реагенты для реакции были предоставлены Шведским ветеринарным университетом (SVA) города Упсала. Для выделения вируса использовали клоакальные смывы и кусочки органов от домашних и диких птиц. До начала исследований пробы хранили в низкотемпературном морозильнике (±70 ºС). Таблица. Результаты исследованных образцов БН в РВ-ПЦР
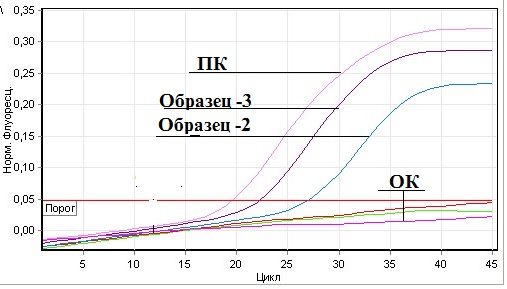
Как видно из таблицы, из 22 исследованных проб в 2-х выявлены РНК вируса болезни Ньюкасла. При этом положительный результат отмечен только у кур. Все пробы уток, голубей, куропаток, индюков из Гиссарского района и диких голубей из джамоата Чимтеппа района Рудаки дали отрицательный результат в РВ-ПЦР. Результаты ПЦР в реальном времени приведены на рис. 1.
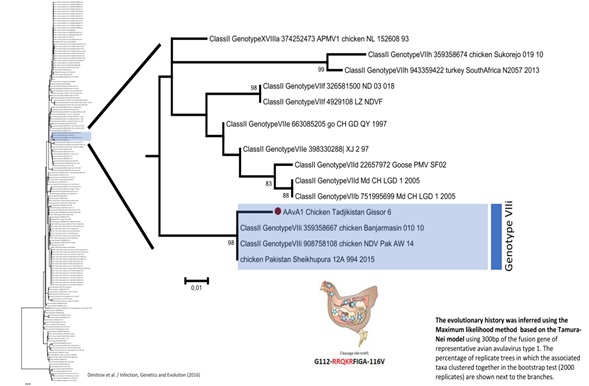
Рис.1. Результаты РВ-ПЦР на вирус БН Совместно со специалистами Шведского национального ветеринарного университета (SVA, Упсала) изучено филогенетическое дерево изолята вируса болезни Ньюкасла, выделенного в Таджикистане. После выделения вируса проведено секвенирование фрагмента гена, кодирующего белок слияния (F-протеин). В результате секвенирования расшифрована последовательность нуклеотидов, которая включает сайт расщепления белка слияния, что важно для выявления его патогенности. Для определения филогенетических взаимоотношений секвенированного вируса осуществлен анализ имеющихся в международной базе данных полных нуклеотидных последовательностей F-гена (GGRRQKRFIGAV) ПМВ-1. Результаты молекулярно-биологического исследования в полимеразной цепной реакции с обратной транскрипцией (ОТ-ПЦР) и методом нуклеинового секвенирования генома позволили идентифицировать изолят от домашних птиц, относящийся к авулавирусу птиц 1- серотипа,7i — генотипа (VIIi), т.е. вирус БН (Avian Avulavirus type 1) (рис. 2).
Рис. 2. Дендрограмма Гиссарского изолята вируса болезни Ньюкасла по антигенному родству выполненная в Шведском национальном ветеринарном университете (SVA), Упсала. Как видно из рис. 2, Гиссарский изолят вируса болезни Ньюкасла (на рисунке текст выделен) по антигенному варианту совпадает на 98 % с штаммами стран южной Азии, такими как Шри Ланка и Пакистан. Заключение Авторы Д. М. Шоназар, кандидат ветеринарных наук, заведующий лабраторией диагностики вирусных болезней животных и птиц Института проблем биологической безопасности ТАСХН (ИПББ), Душанбе Библиографические ссылки Читайте также:
 Пожалуйста, не занимайтесь самолечением!При симпотмах заболевания - обратитесь к врачу. Пожалуйста, не занимайтесь самолечением!При симпотмах заболевания - обратитесь к врачу.
Copyright © Иммунитет и инфекции
|