
Что такое секвенирование вируса
Исследователи из Public Health England опубликовали на BioRxiv протокол для метагеномного секвенирования РНК-вирусов из клинических образцов без амплификации или обогащения. Протокол протестирован на секвенаторах Oxford Nanopore MinIon и Illumina MiSeq.

Подготовила Елена Клещенко
Метагеномное секвенирование широко используется в исследовании инфекционных агентов, диагностике и эпидемиологии. Стивен Пуллен, руководитель проекта по геномике группы редких и новых человеческих патогенов в Public Health England (исполнительного органа департамента здравоохранения и соцобеспечения Великобритании), заявил, что протокол аналогичен стратегии метагеномного секвенирования для MinIon, описанной группой Чарльза Чиу (Калифорнийский университет в Сан-Франциско) в 2015 году. Авторы работы показали , что MinIon может идентифицировать патоген (вирус чикунгунья, лихорадки Эбола и гепатита С) в крови. Однако они исследовали всего несколько образцов с высокой вирусной нагрузкой, тогда как работа PHE была проведена на многих образцах с различными нагрузками. Как отмечает Пуллен, они стремились создать протокол, подходящий для полевой работы на месте вспышки заболевания, когда нет времени и условий для культивирования патогена.
Сотрудники PHE сфокусировались на РНК-вирусах лихорадки денге и чикунгунья, отчасти потому, что у них был доступ к образцам пациентов британской Лаборатории редких и импортных патогенов. Так, они исследовали 26 образцов, положительных по вирусам денге или чикунгунья. Одним из ключевых этапов протокола Пуллен назвал деградацию ДНК после экстракции нуклеиновых кислот, — удаление ДНК облегчает идентификацию РНК-вируса. После этого команда подготовила кДНК с использованием стратегии сиквенс-независимой амплификации (аналогичной той, что была описана командой Чиу), а затем выполнялись случайная обратная транскрипция и секвенирование.
Примечательно, что хотя секвенирование на Illumina давало больший процент ридов из вирусных геномов, секвенирование с 20-кратным покрытием на MinIon обеспечило аналогичное покрытие генома: например, даже в образце с вирусом чикунгунья, для которого получили самый низкий процент вирусных чтений (5% на MinIon и 22% на MiSeq), при 20-кратном покрытии с MinIon было просеквенировано более 89% генома.
В целом, сказал Пуллан, как MinIon, так и MiSeq позволяли идентифицировать вирус и получить последовательность большей части вирусного генома. Кроме того, исследователи определили в одном из образцов коинфекцию вирусами чикунгунья и денге. Хотя только 0,08% ридов MiSeq и 0,15% ридов от MinIon соответствовали геному вируса денге, 20-кратное секвенирование на MinIon охватило как первичный вирус чикунгунья, так и вирусы денге более чем на 99% и 95% соответственно.
Во время исследования Oxford Nanopore выпустил два новых комплекта для подготовки библиотек — комплект 1D 2 и быстрый комплект , — которые также испытали сотрудники PHE. Хотя при использовании обоих комплектов доля вирусных ридов снизилась, 1D 2 увеличил объем данных до 5 миллионов чтений (при 1,8 миллиона у 2D, т.е. секвенировании обеих нитей). Преимущество Rapid Sequencing Kit от Oxford Nanopore — ускоренная и упрощенная подготовка образца всего за 10 минут.
По словам Пуллена, команда планирует продолжить тестирование различных метагеномных протоколов, чтобы выяснить, какие технологии лучше подходят для тех или иных приложений. Сейчас они уделяют основное внимание протоколам для полевой работы, для отслеживания вспышек эпидемий в режиме реального времени, но также будет рассмотрен вопрос о разработке диагностического теста на вирусы денге или чикунгунья. Кроме того, группа участвует в проектах по разработке метагеномных диагностических анализов на грипп из респираторных образцов.
Liana E Kafetzopoulou et al. // Assessment of Metagenomic MinION and Illumina sequencing as an approach for the recovery of whole genome sequences of chikungunya and dengue viruses directly from clinical samples. // BioRxiv 2018 DOI: 10.1101/355560
Полное представление о том, с какими разновидностями вируса приходится иметь дело при начале мероприятий по обузданию инфекции и борьбе с ней является критичным как для эффективного их мониторинга, так и для идентификации новых, интродуцированных на фермах или в регионе, штаммов.
Геном вируса репродуктивно-респираторного синдрома свиней (РРССВ) представлен одноцепочечной молекулой РНК, что объясняет его крайнюю подверженность генетическим мутациям. Это также делает каждый штамм вируса РРСС уникальным - настолько, что генотипирование является весьма полезным методом диагностики и борьбы с заболеванием. Диагностическое генотипирование проводится путем определения последовательности нуклеотидов на отрезке ДНК, копирующем геномный фрагмент вирусной РНК (секвенирование ДНК). Наиболее часто для этого в настоящее время используется фрагмент ORF5 – ген, кодирующий основной оболочечный гликопротеин. В значительной мере это обусловлено тем, что он обладает неограниченным генетическим разнообразием.

Диагностическое секвенирование ДНК
Разница между вирусами РРСС первого (европейский) и второго (американский) типа легко выявляется с помощью большинства диагностических ПЦР-тест-систем. В то же время, установление различий между отдельными штаммами каждого из этих двух генотипов требует проведения секвенирования ДНК. С этой целью биологические жидкости или ткани, содержащие вирус РРСС в умеренных или высоких концентрациях, обрабатываются таким образом, чтобы изолировать РНК, которая затем копируется на ДНК методом обратной транскрипции, после чего ген ORF5 амплифицируется с помощью ПЦР и попадает на оборудование для проведения секвенирования ДНК. Этот процесс почти полностью автоматизирован и обычно занимает от одного до трех дней. Полученные в результате необработанные данные поступают в диагностическую лабораторию для анализа. Стандартный отчет о результатах содержит описание нуклеотидных последовательностей исследованного штамма, его сходство со стандартными штаммами, используемыми в вакцинах. Некоторые лаборатории, помимо этого, предоставляют результаты сравнения со стандартными панелями изолятов вируса РРСС дикого типа, представленные в виде дендрограмм. Также, могут предоставляться данные о результатах сравнения с последовательностями РРССВ, описания которых содержатся в производственной базе данных.
Анализ последовательностей РРССВ
Схожесть или идентичность последовательностей выявляется путем сопоставления двух или более последовательностей с помощью компьютерной программы. Пример сопоставления нескольких последовательностей ORF5 с пяти различных ферм приведен на Рис.1. Попарные сравнения процентной степени их идентичности, лежащие в пределах от 81,2% до 99,8%, представлены в таблице. Дендрограмма результатов полигенетического анализа демонстрирует группировку схожих последовательностей (Рис.2). Ключевой вопрос, встающий перед производителями и ветеринарами, это – являются ли выявленные генетические различия в последовательностях обычными вариациями одного и того же штамма вируса РРСС или они свидетельствуют о присутствии на ферме различных штаммов?

Рис.1. Фрагмент сопоставления последовательностей ORF5 штаммов вируса РРСС пяти разных ферм. С ферм 1, 4 и 5 было получено по нескольку последовательностей. Точки в последовательностях соответствуют позициям, идентичным референтному штамму Lelystad вируса РРСС первого типа.
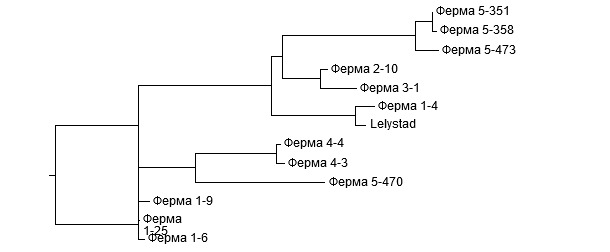
Рис.2. Дендрограмма последовательностей ORF5, полученных для пяти разных ферм. Пример расшифровки: на Ферме 1 присутствуют 2 несвязанных штамма. Степень идентичности трех последовательностей друг другу >99%, в то время, как четвертая соответствует им только на
83%. Зато она соответствует вирусу Lelystad. Два штамма с Фермы 4 тесно связаны друг с другом (идентичны на 99,5%). На Ферме 5 присутствуют два несвязанных штамма. Степень соответствия трех штаммов друг другу >98%, а четвертому – только около 81%.
Таблица. Попарное представление идентичности всех сопоставленных последовательностей ORF5 серий образцов вируса РРСС первого типа в процентах.
| 1-4 | 1-6 | 1-9 | 1-25 | 2-10 | 3-1 | 4-4 | 4-3 | 5-351 | 5-358 | 5-470 | 5-473 | Lelystad | |
| *** | 83.5 | 83.3 | 83.7 | 93.2 | 92.6 | 86.1 | 86.0 | 88.1 | 88.0 | 83.2 | 88.3 | 98.7 | 1-4 |
| *** | 99.2 | 99.7 | 84.2 | 83.0 | 86.0 | 86.0 | 81.4 | 81.2 | 86.3 | 81.4 | 82.1 | 1-6 | |
| *** | 99.5 | 84.5 | 83.3 | 85.6 | 85.6 | 81.4 | 81.2 | 86.5 | 81.4 | 81.9 | 1-9 | ||
| *** | 84.5 | 83.3 | 86.0 | 86.0 | 81.7 | 81.5 | 86.8 | 81.7 | 82.2 | 1-25 | |||
| *** | 98.2 | 86.6 | 86.5 | 91.1 | 90.9 | 84.2 | 90.4 | 93.3 | 2-10 | ||||
| *** | 84.4 | 84.2 | 90.2 | 90.0 | 83.3 | 89.7 | 92.9 | 3-1 | |||||
| *** | 99.5 | 82.5 | 82.7 | 90.6 | 82.5 | 84.8 | 4-4 | ||||||
| *** | 82.3 | 82.5 | 90.9 | 82.2 | 84.6 | 4-3 | |||||||
| *** | 99.8 | 81.0 | 98.3 | 88.4 | 5-351 | ||||||||
| *** | 81.2 | 98.2 | 88.2 | 5-358 | |||||||||
| *** | 80.9 | 83.2 | 5-470 | ||||||||||
| *** | 88.8 | 5-473 | |||||||||||
| *** | Lelystad |
Интерпретация последовательностей РРССВ
Последовательность ORF5 содержит около 600 нуклеотидов. Общая мутируемость этого гена, по разным оценкам, составляет от 0,5% до 1% в год. Отличия в скорости генетических изменений определяются различными факторами невирусной природы. На репликацию и трансмиссивность вируса РРСС у свиней большое внимание оказывает уровень их специфического и неспецифического иммунитета, создающий сильное ингибирующее давление, приводящее к уменьшению числа вирусных копий. Уменьшение вирусной нагрузки приводит к снижению трансмиссивности, снижая тем самым общую репликацию вируса и степень его изменчивости. Таким образом, иногда может наблюдаться более высокая или более низкая генетическая изменчивость, выходящая за предполагаемые рамки 0,5%-1% в год.
Главным вопросом при проведении генетического анализа является вопрос о том, являются ли две последовательности близкородственными (двумя вариантами одного и того же штамма) или независимыми (принадлежащим двум несвязанным штаммам). Общепринято, что вывод о том, связаны ли изоляты вируса РРСС или нет, делается, исходя из степени схожести, попадающей в интервал достоверности 97% или 98%. Очевидно, что безоговорочное, сделанное без учета всех сопутствующих факторов, принятие за основу предпосылки о двух- или трехпроцентной генетической разнице между двумя изолятами может привести к неверным выводам. Различия между двумя вариантами одного и того же штамма, циркулирующего в поголовье на протяжении нескольких лет, постепенно выбьются за пределы данного интервала. Степень достоверности выводов о близкородственности может быть повышена получением доступа к дополнительной информации, включая даты и локализацию получения изолятов. Очень важен анализ, противопоставляющий новые последовательности РРССВ широкому референтному набору, являющемуся показательным для фермы, региона, используемой технологии или даже генетического разнообразия в глобальном масштабе.
Секвенирование ДНК штаммов вируса РРСС может вскрыть их родственность или независимость (Рис.2), но не может быть использовано для прогноза или объяснения степени иммунологической защиты или вспышек в иммунных поголовьях. Также, секвенирование ДНК не позволяет судить о клинических последствиях инфицирования данным конкретным штаммом, в силу того, что идентификация генетических маркеров вирулентности отсутствует.
В настоящее время, в основном в США, но также и в Европе осуществляется множество региональных проектов по борьбе с РРСС. Полное представление о том, с какими разновидностями вируса приходится иметь дело при начале мероприятий по обузданию инфекции и борьбе с ней является критичным как для эффективного их мониторинга, так и для идентификации новых, интродуцированных на фермах или в регионе, штаммов.
ТЕХНОЛОГИИ СЕКВЕНИРОВАНИЯ НОВОГО ПОКОЛЕНИЯ (NGS) ДЛЯ ИССЛЕДОВАНИЯ МИКРООРГАНИЗМОВ
Применение секвенирования нового поколения (NGS) в микробиологии позволяет достигнуть нового уровня в понимании того, как микробы воздействуют на людей и окружающую среду.
Благодаря высокой производительности секвенаторов Illumina теперь Вы можете изучить генетическое строение организмов, которые ранее изучить было невозможно.

16S-метагеномика ― это широко известный метод секвенирования, используемый для идентификации и сравнения бактерий, присутствующих в изучаемом образце, и определения штаммов, которые не могут быть обнаружены другими методами.

Шотган-метагеномика обнаруживает даже малочисленных членов микробного сообщества, которые могут быть пропущены другими методами, благодаря возможности секвенирования множества образцов микроорганизмов за запуск и высокой степени покрытия на образец.

В отличие от методов, основанных на гибридизации, таких как ДНК-биочипирование, секвенирование РНК микроорганизмов делает возможной цепь-специфическую идентификацию как распространённых, так и новых транскриптов.

Полногеномное секвенирование (WGS) методом NGS предоставляет Вам возможность мультиплексного секвенирования сотни организмов. И, в отличие от традиционных методов, не требуется трудоемких этапов клонирования.
АНАЛИЗ МИКРОБИОМА ЧЕЛОВЕКА
Анализ микробиома человека ― это изучение микробных сообществ, обнаруженных в организме человека и на нём. Целью исследований микробиома человека является понимание роли микробов в его здоровье и заболеваниях.
Традиционно изучение образцов кожи, кала или крови человека основывалось на трудоёмких микробиологических техниках выращивания и выделения отдельных организмов, за которыми следовал фенотипический или генотипический анализ. Этими методами было невозможно профилировать микробные сообщества в рамках одного образца.
МЕТАГЕНОМИКА ОКРУЖАЮЩЕЙ СРЕДЫ
Метагеномика окружающей среды ― это изучение микроорганизмов на основе анализа ДНК в образцах окружающей среды.

Примеры включают профилирование популяций микроорганизмов в образцах воды, взятых из глубоких океанских впадин или в образцах почвы из среды, преобразованной в следствии действий человека, таких, например, как активные добычи минералов. Данные исследования метагеномики окружающей среды используются для анализа сельскохозяйственных микробиомов, востановления экологии или других биологических исследований.
Метагеномика окружающей среды как исследовательское поле была крайне ограничена до появления секвенирования нового поколения (NGS). NGS предоставляет исследователям возможность профилировать любые микробные сообщества, открывать новые организмы и исследовать микробные популяции в динамике..
НАДЗОР ЗА РАСПРОСТРАНЕНИЕМ ИНФЕКЦИОННЫМИ ЗАБОЛЕВАНИЯМИ
Геномика расширяет наше понимание эволюции патогенов, взаимодействия хозяин-патоген и устойчивости к антибиотикам. Геномика трансформирует современный эпиднадзор за инфекционными заболеваниями и методы, с помощью которых специалисты общественного здравоохранения работают над защитой от эпидемий и пандемий массово распространённых заболеваний, таких как туберкулез, ВИЧ и грипп.
Современные методы изучения возбудителей инфекционных заболеваний включают тестирование на основе антител, real-time PCR, гель-электрофорез в пульсирующем поле (PFGE) и мультилокусное секвенирование (MLST). Эти методы обычно применимы для изучения только небольшого количества определенных организмов, и анализ данных может быть субъективным.
Секвенирование нового поколения (NGS) ― это универсальный метод, который можно использовать при изучении и вирусов, и бактерий, и паразитов. NGS может заменить сразу несколько методов и предоставляет высокоточные данные, позволяя различать штаммы патогенов, которые отличаются всего на один SNP.
Технология NGS от Illumina позволяет быстро получить данные высокого качества, которые специалисты по инфекционным заболеваниям будут использовать для выявления, отслеживания и реагирования на вспышки.
Секвенирование полных геномов микроорганизмов является основным методом эпиднадзора за инфекционными заболеваниями.

МОЛЕКУЛЯРНАЯ ЭПИДЕМИОЛОГИЯ

Молекулярная эпидемиология позволяет изучать генетическую структуру популяций микроорганизмов для выяснения причин возникновения эпидемически-значимых патогенов, путей его распространения и передачи, источник инфекции. Она также используется для установления генов, ответственных за вирулентность и лекарственную устойчивость.
Традиционные способы идентификации и исследования патогенов основаны в основном на методе последовательных гипотез и подходят для изучения ограниченного числа организмов.
Секвенирование нового поколения (NGS) осуществляет высокоточный анализ множественных изолятов, может заменять несколько тестов для идентификации организма и изучения лекарственной устойчивости и вирулентности одновременно. Высокоточное типирование патогенов с использованием данных полногеномного секвенирования может различать организмы, которые многие современные методы различить не могут.
Ключевым преимуществом NGS является то, что исследователи могут получать данные секвенирования генома, способные идентифицировать ряд организмов (бактерии, вирусы и паразиты). Изучение вариаций в геномах проводится методом кросс-секционного, межгруппового исследования поперечного сечения для лучшего понимания механизмов, которые приводят к инфекции и её распространению, а также филогенетического анализа для определения родства между организмами.
Основным методом молекулярной эпидемиологии является секвенирование полных геномов микроорганизмов.
ИССЛЕДОВАНИЯ МИКРОБИОМА ДЛЯ ЛЕЧЕНИЯ РАКА
Микробы, живущие в человеке, могут влиять на прогрессирование рака и эффективность лечения. Диета и лекартсва могут нарушать многообразие микробиома, а определённые микроорганизмы в микробиоме могут вызывать локальные или системные воздействия на иммунитет хозяина.
Мы надеемся, что в будущем лечение может сочетать существующие методы лечения рака с методами поддержания полезных микробов или устранения вредных. Поскольку исследования, основанные на NGS, продолжают исследовать взаимодействия хозяина и его микробиома, Illumina разрабатывает геномные технологии в этой области.
Методы NGS произвели революцию в изучении микробиома. При использовании NGS не нужно проводить культивирование или клонирование отдельных организмов. NGS позволяет проводить одновременный анализ тысяч видов в микробном сообществе. В дополнение к этому, развитие биоинформатических инструментов для управления большими объемами новых данных позволяет точно оценить видовое разнообразие и измерение динамических колебаний микробных сообществ.
Расшифровка генома какао — одно из самых громких научных достижений в генетике за последнее время. Итоги двух лет исследований ученые представили в Цюрихе журналистам, среди которых был и корреспондент "Огонька"
Кирилл Журенков, Цюрих — Москва
"Ведьмина метла", или "эскоба де бруха",— это название приводит в ужас, пожалуй, всех фермеров Южной Америки. Вредный грибок не только уничтожает плоды и листву, но и проникает в стволы деревьев. Как результат: целые плантации зараженного какао. Считается, что во многом именно из-за этой болезни Южная Америка, которая некогда была лидером по производству какао, уступила свое почетное место другому континенту — Африке.
И вот сегодня ученые обещают: "ведьмина метла" и другие подобные болезни, наносящие огромный ущерб сельскому хозяйству по всему миру, скоро уйдут в прошлое. На помощь фермерам пришла генетика, точнее — новейшие достижения в области расшифровки генома.
В международном проекте по секвенированию (именно так правильно называть определение последовательности нуклеотидов в молекуле ДНК) генома какао приняло участие множество самых различных организаций: компания "Mapc", известная в России по шоколадкам, IBM, Минсельхоз США, Университет Калифорнии, Государственный университет Вашингтона, Университет Индианы — и это лишь начало списка.
Об успешном завершении исследований было объявлено несколько месяцев назад, а недавно один из руководителей проекта доктор Говард-Яна Шапиро представил результаты секвенирования журналистам на встрече в исследовательской лаборатории IBM в Цюрихе. Формат видеоконференции, когда вопросы ученому задавали в Цюрихе, а отвечал он на них из офиса в США, еще раз продемонстрировал полную победу науки над расстояниями, а заодно и над природой. Что касается самого участия всемирно известной корпорации, то оно не случайно: секвенирование генома требует нешуточных компьютерных мощностей.
— Ключевое слово здесь не биология, а информатика,— говорит Станислав Полонский, руководящий одним из направлений по исследованию генома в компании IBM.— Можно продемонстрировать какие-то алгоритмы, применить новые научные подходы. Так почему бы нет?
Вся эта команда объединилась, чтобы ни много ни мало спасти индустрию производства шоколада. Дело в том, что деревья какао, из плодов которого сегодня и производят шоколад,— одни из самых уязвимых растений на планете. Вот статистика: каждый год только западноафриканские производители какао теряют до 800 млн долларов из-за всевозможных вредителей и плохих погодных условий. А по данным Института природных ресурсов графства Кент (Великобритания), за последние годы от болезней погибло почти 60 процентов урожая какао в Бразилии.
Всего же в мире выращивают эту культуру примерно 6,5 млн фермеров, и для них борьба за урожай — зачастую вопрос выживания. Важность проекта исследователи объясняют именно этим: какао сегодня кормит множество людей по всей планете.
— Нам понадобилось два года, два месяца и 20 дней, хотя мы думали, что это займет лет пять,— сказал "Огоньку" доктор Говард-Яна Шапиро из компании "Марс".
Пока что представленные результаты охватывают 92 процента всего генома какао — это приблизительно 35 тысяч генов (для сравнения: у человека около 20 тысяч генов). Однако исследователи обещают, что завершат начатое, а о результатах будут сообщать на специально созданном веб-сайте. "Вся информация будет доступна каждому",— уверяет доктор Шапиро. Сегодня геном уже стал общественным достоянием.
Теперь, как считают исследователи, появляется возможность улучшать характеристики растения на генетическом уровне, а значит, повышать урожайность, устойчивость к паразитам, качество самого какао. Это сулит и социальные преимущества: увеличение дохода фермеров, улучшение экологии (исчезнет нужда в химикатах, сами плантации будут занимать меньше земли). Доктор Шапиро видит основания говорить о новой "зеленой революции".
Геном за миллион
Секвенирование генома какао — значимое, но далеко не единственное подобное достижение в генетике. В последние годы ученых со всего мира охватила настоящая лихорадка: расшифровывают геномы всех живых организмов — от бактерий до человека. А несколько лет назад американский фонд X Prize, присуждающий премии за передовые научные исследования, даже объявил вознаграждение в 10 млн долларов за создание самого быстрого метода секвенирования. Условия получения гранта довольно жесткие, но сегодня они уже не кажутся невозможными: 100 произвольных человеческих геномов за 10 дней, при этом стоимость исследований должна быть не выше 10 тысяч долларов за геном.
Биолог, заместитель директора по научным вопросам Института проблем передачи информации РАН Михаил Гельфанд вспоминает, что начиналось все в 1980-х годах с секвенирования вирусов. Сейчас такие исследования, в частности, помогли в борьбе с атипичной пневмонией: определение последовательности вируса позволило установить, к какой группе он принадлежит, и понять механизмы заражения.
Затем ученые обратили внимание на бактерии, а после бактерий сразу схватились за самый амбициозный эксперимент — расшифровку человеческого генома. Здесь практических шагов также множество: от специальных тестов, позволяющих производить раннюю диагностику рака, до развития так называемой индивидуальной медицины (то есть подбора лекарств непосредственно под геном конкретного человека).
К 2000-м, когда вспомнили, наконец, о растениях, человеческий геном был уже практически расшифрован. Одной из причин, по которой с этим не торопились, ученые называют то, что геномы растений определить гораздо сложнее — многие из них длиннее, чем у человека. Однако развитие технологий и удешевление процесса секвенирования сделали возможным и это.
Впрочем, дело, конечно, не только в научной гонке. Как подчеркивают сами ученые, они в тревоге за судьбу планеты.
Первым сельскохозяйственным растением, геном которого был секвенирован, стал рис — базовая культура питания для большей части населения планеты. Вслед за ним благодаря тому, что технологии совершенствовались, были расшифрованы геномы и других важнейших сельскохозяйственных культур: картофеля, кукурузы, пшеницы. Собственно, сегодня последовательность нуклеотидов в молекуле ДНК определена для большинства растений, которые являются основой рациона питания.
Но останавливаться еще рано. По словам одного из исследователей генома пшеницы, Майка Бевана из Центра Джона Иннса (этот независимый международный центр занимается микробиологией и науками о растениях), наука просто не успевает давать ответы на вопросы, которые ставит растущий дефицит продовольствия.
Впрочем, у "генетической гонки" есть и другая сторона.
— Сегодня технологии секвенирования шагнули далеко вперед и сделали этот метод рентабельным. Иными словами, на нем можно сделать хороший бизнес,— говорит Сергей Киселев, директор отделения нейронауки и молекулярно-клеточной биологии Курчатовского института.— Если говорить о секвенировании растений, речь идет о тех видах, которые мы используем на протяжении сотен тысяч лет: рисе, пшенице, сое или хлопке — обо всем, что находится в коммерческом обороте и дефицит чего, как утверждают в последнее время, мы стали ощущать все сильнее. Соответственно, зная те или иные геномы, можно создать ускорители роста или изобрести принципиально новые средства борьбы с болезнями этих растений.
По словам Киселева, трансгенные растения, из которых сегодня производят продукты,— результат первых работ по секвенированию, когда были вычислены только части геномов, а не вся цепочка ДНК. В будущем же, утверждают специалисты, необходимость в трансгенах вообще отпадет. Напротив, как сегодня уже делается в медицине, люди получат возможность контролировать биологические процессы, что называется, по ходу дела: к примеру, будет достаточно посыпать особым порошком плантацию риса, и его рост ускорится, чтобы успеть к заморозкам.
Не случайно только в Китае Пекинским институтом геномных исследований в начале года было закуплено примерно 130 самых высокопроизводительных аппаратов для установления цепочек ДНК (стоимость подобного устройства — порядка 400 тысяч долларов): по данным на минувшее лето, с их помощью было секвенировано около 300 геномов растений.
Какое место займет в этой гонке наша страна — вопрос пока открытый.
— В России, безусловно, занимаются секвенированием, но отечественная наука явно не на лидирующих позициях,— объясняет Станислав Полонский из IBM.
Сергей Киселев из Курчатовского института лишь пожимает плечами: изучение генома растений для российских ученых вопрос интересный, но не слишком актуальный. Зато есть другие важные темы, например здоровье нации. Секвенирование генома раковых опухолей и вообще изучение генетических особенностей тех или иных заболеваний для создания новых лекарств, по мнению Киселева, действительно насущная задача для российских ученых.
С геномом на ты

Фото: Science Photo Library/Eastnews
Что же касается традиционных опасений, связанных с вмешательством в геном, то их ученые отметают с порога.
— Не мы одни экспериментируем — природа экспериментирует вместе с нами,— говорит Станислав Полонский.— Возьмите каждое новое поколение вирусов или микробов — там идут постоянные генетические модификации. Конечно, если говорить о генетических модификациях, то здесь можно проколоться, но, с другой стороны, разве это плохо, когда в рис встраивают витамины, чтобы восполнить их недостаток у людей в тех или иных странах? Родятся ли от этого люди-мутанты? Честно говоря, сомневаюсь. Но вообще это разговор о прогрессе и его возможных издержках: на свет можно ненароком произвести и нечто ужасное, но скорее всего плюсов будет все равно больше.
Михаил Гельфанд из Института проблем передачи информации РАН также оптимистично смотрит в будущее. По его словам, эти опасения — плод невежества.
— Это вовсе не означает, что не надо проверять на безопасность и жестко лицензировать сорта генно-модифицированных растений, и, конечно, ученые отвечают за результат вместе с производителями,— уточняет он.— Но подобные меры предосторожности нужны при работе с любой новой технологией от пластмасс, удобрений и прочих продуктов химической промышленности до персональных компьютеров и сотовых телефонов.
Открытия в генетике врываются в жизнь так стремительно и меняют ее так решительно, что далеко не все мы успеваем это понять. Ученым легче: тот же доктор Шапиро объясняет, что, когда он был ребенком, одной из самых страшных болезней в мире считался полиомиелит (от него страдал и один из друзей исследователя). В результате человечеству помогла открытая генетиками вакцина Сэбина.
— Мне тогда было 5 лет, я не знал про эту вакцину, и кто-то принял решение ее использовать за меня,— разводит руками доктор Шапиро.— Это было рискованно, но это спасло миллионы детей по всему миру.
За последние годы секвенированы геномы основных сельскохозяйственных растений
Рис стал первой сельскохозяйственной культурой, чей геном был расшифрован, хотя самое первое растение в мире, геном которого был секвенирован в 2000 году,— сорняк Arabidopsis. Расшифровка генома риса закончена в декабре 2004 года: установлено, что в нем содержится 37 544 генов. В исследованиях приняли участие ученые из 10 стран. Ожидалось, что работы затянутся лет на десять, но их завершили за шесть.
О завершении черновой расшифровки генома (32 тысячи генов) этого растения было сообщено в начале 2008-го. Руководили исследованиями ученые из Вашингтонского университета в Сент-Луисе. Проект обошелся в 29,5 млн долларов, полученных от Национального научного фонда, Министерства сельского хозяйства и Министерства энергетики США.
Геном в черновом варианте расшифрован в сентябре прошлого года. Число генов не сообщается, зато известно, что он состоит из 840 млн пар нуклеотидов (геном яблока, для сравнения, содержит в себе 600 млн пар нуклеотидов). Участие в секвенировании принимали 16 групп ученых из разных стран, в том числе России.
Черновой вариант расшифрованного генома опубликован в августе этого года. В нем содержится 80 тысяч генов — это самый большой из известных геномов. Над секвенированием работали специалисты Университета Ливерпуля, а заняло это у них всего лишь год.
Читайте также:
