Инфекция мочевыводящей системы
Наиболее распространенным симптомом ПБЦ является усталость (повышенная утомляемость), которая возникает у 70% больных. Следует отметить, что значительная усталость может быть причиной нарушения сна или депрессивного состояния.
Заболевание начинается внезапно, чаще всего с зуда кожи на ладонях рук и подошвах ног, не сопровождающегося желтухой. Позже он может распространиться на все тело. Интенсивность кожного зуда может усиливаться ночью и уменьшаться в течение дня. Ночной зуд часто нарушает сон и приводит к усилению усталости. Причина развития зуда на сегодняшний день остается неясной.
Примерно у 25% больных ПБЦ на момент постановки диагноза выявляют ксантомы. Появлению ксантом предшествует длительное (более 3 месяцев) повышение уровня холестерина в крови более 11.7 мкмоль/л.
Разновидность ксантом — ксантелазмы — плоские или слегка возвышающиеся мягкие безболезненный образования желтого цвета, обычно располагающиеся вокруг глаз. Но ксантомы могут так же наблюдаться в ладонных складках, под молочными железами, на шее груди или спине. Они исчезают при разрешении (исчезновении) холестаза и нормализации уровня холестерина, а так же при развитии конечной стадии заболевания (печеночной недостаточности) в связи с нарушением синтеза холестерина в поврежденной печени.
Так же при хроническом холестазе ПБЦ (в связи с нарушенным выделением желчи) развивается нарушение всасывания жиров и жирорастворимых витаминов (A, D, Е и К), что может привести к диарее, потере веса и недостатку данных витаминов.
Дефицит витамина А вызывает снижение зрения в темноте. Дефицит витамина Е может проявляться неприятными ощущениями на коже или мышечной слабостью. Дефицит витамина D способствует прогрессированию костных изменений (остеомаляции, остеопороза). Дефицит витамина К приводит к снижению синтеза печенью белков свертывающей системы и, следовательно, к склонности к кровотечениям.
Диагностика ПБЦ
- Общеклинические анализы крови - оценка работы печени, напряженности иммунитета, а так же определение показаний к лечению.
- Исключение других заболеваний печени, протекающих с хроническим холестазом — в том числе инструментальными методами (УЗИ, МРХПГ, эндоскопическая ультрасонография и т.д.).
- Анализ крови на аутоантитела (АМА и другие).
- Фибротест — анализ крови, назначающийся при подозрении на билиарный цирроз, позволяющий оценить степень воспалительных и фибротических изменений в ткани печени.
- Инструментальные методы диагностики (УЗИ органов брюшной полости, ФГДС и т.д.) - для оценки состояния гепатобилиарной системы и выявления возможных осложнений (в том числе признаков цирроза).
- При необходимости - биопсия печени с гистологическим исследованием —для исключения синдромов перекреста с другими заболеваниями печени, подтверждения диагноза, определения стадии заболевания и степени повреждения печени (в том числе наличие цирроза).
При отсутствии типичных для первичного билиарного цирроза аутоантител (АМА), наличии типичной клинической сипмтоматики и типичных для первичного билиарного цирроза изменений печени по данным биопсии — диагностируют АМА-негативный ПБЦ или так называемый Аутоиммунный холангит.
Лечение
- Урсодезоксихолевая кислота (УДХК) – доказанное эффективное лечение ПБЦ. При отсутствии ответа на УДХК к терапии добавляют иммуносупрессоры.
- Для уменьшения кожного зуда пациентам рекомендуется не носить синтетическую одежду (используйте хлопок, лен), избегать горячих ванн и перегревания, всегда держать ногти коротко остриженными. Можно использовать прохладные ванны с содой (чайная чашка на ванну) по горло 20 минут, ножные ванны с содой. Из препаратов, способных уменьшит зуд, используют холестирамин, рифампицин, при их неэффективности - пероральные антагонисты опиатов и сертралин. Так же в ряде случаев положительный эффект на зуд может оказать плазмаферез.
- Профилактика дефицита жирорастворимых витаминов заключается в полноценном питании и компенсации стеатореи (потери жира с калом) ферментативными препаратами.
- С целью профилактики развития остеопороза врач может назначить препараты, содержащие кальций и витамин D.
- Трансплантация печени остается единственным методом лечения при декомпенсации цирроза, при инвалидизирующем зуде и выраженном остеопорозе - 10-летняя выживаемость при ПБЦ после трансплантации составляет около 70 %.
Прогноз
Течение первичного билиарного цирроза при отсутствии симптомов непредсказуемо, в отдельных случаях симптомы не развиваются вообще, в других отмечается прогрессирующее ухудшение с развитием цирротической стадии. Часто причиной смерти при циррозе является кровотечение из варикозно-расширенных вен пищевода и желудка.
А в терминальной стадии пациенты погибают от развития печеночной недостаточности.
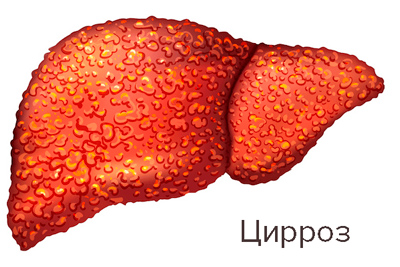
Исходом хронического холестаза и воспаления в печени, является цирроз печени.
При условии эффективности и соблюдения лечения большинство людей с первичным билиарным циррозом имеют нормальную продолжительность жизни.
![]()
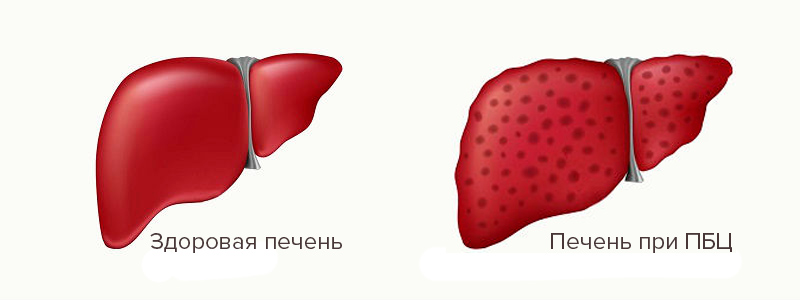
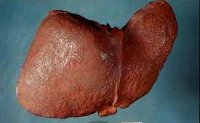
Первичный билиарный цирроз печени – хронический прогрессирующий деструктивно-воспалительный процесс аутоиммунного генеза, поражающий внутрипеченочные желчные протоки и приводящий к развитию холестаза и цирроза. Первичный билиарный цирроз печени проявляется слабостью, кожным зудом, болью в правом подреберье, гепатомегалией, ксантелазмами, желтухой. Диагностика включает исследование уровня печеночных ферментов, холестерина, антимитохондриальных антител (АМА), IgM, IgG, морфологическое исследование биоптата печени. Лечение первичного билиарного цирроза печени требует проведения иммуносупрессивной, противовоспалительной, антифибротической терапии, приема желчных кислот.
![]()
Общие сведения
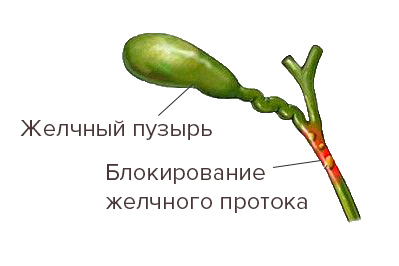
Первичный билиарный цирроз печени развивается преимущественно у женщин (соотношение заболевших женщин и мужчин 10:6), средний возраст пациентов составляет 40-60 лет. В отличие от вторичного билиарного цирроза печени, при котором имеет место обструкция внепеченочных желчных протоков, первичный билиарный цирроз протекает с постепенным разрушением внутрипеченочных междольковых и септальных желчных протоков. Это сопровождается нарушением желчевыделения и задержкой токсических продуктов в печени, приводя к прогрессирующему снижению функциональных резервов органа, фиброзу, циррозу и печеночной недостаточности.
![]()
Причины первичного билиарного цирроза печени
Этиология первичного билиарного цирроза печени неясна. Заболевание часто носит семейный характер. Отмеченная связь между развитием первичного билиарного цирроза печени и антигенами гистосовместимости (DR2DR3, DR4, В8), характерными для патологии аутоиммунного характера. Данные факторы указывают на иммуногенетическую составляющую заболевания, обусловливающую наследственную предрасположенность.
Первичный билиарный цирроз печени протекает с системным поражением эндокринных и экзокринных желез, почек, сосудов и довольно часто сочетается с сахарным диабетом, гломерулонефритом, васкулитом, синдромом Шегрена, склеродермией, тиреоидитом Хашимото, ревматоидным артритом, системной красной волчанкой, дерматомиозитом, целиакией, миастенией, саркоидозом. Поэтому первичный билиарный цирроз печени находится в фокусе внимания не только гастроэнтерологии, но и ревматологии.
В развитии первичного билиарного цирроза печени не исключается пусковая роль бактериальных агентов и гормональных факторов, инициирующих иммунные реакции.
Стадии первичного билиарного цирроза печени
В соответствии с происходящими гистологическими изменениями выделяют 4 стадии первичного билиарного цирроза печени: дуктальную (стадию хронического негнойного деструктивного холангита), дуктуллярную (стадию пролиферации внутрипеченочных протоков и перидуктального фиброза), стадию фиброза стромы и стадию цирроза.
Дуктальная стадия перевичного билиарного цирроза печени протекает с явлениями воспаления и деструкции междольковых и септальных желчных протоков. Микроскопическая картина характеризуется расширением портальных трактов, их инфильтрацией лимфоцитами, макрофагами, эозинофилами. Поражение ограничено портальными трактами и не распространяется на паренхиму; признаки холестаза отсутствуют.
В дуктуллярной стадии, соответствующей пролиферации холангиол и перидуктальному фиброзу, отмечается распространение лимфоплазмоцитарной инфильтрации в окружающую паренхиму, снижение числа функционирующих внутрипеченочных протоков.
В стадии фиброза стромы на фоне воспаления и инфильтрации печеночной паренхимы отмечается появление соединительнотканных тяжей, соединяющих портальные тракты, прогрессирующая редукция желчных протоков, усиление холестаза. Происходит некроз гепатоцитов, нарастают явления фиброза в портальных трактах.
В четвертой стадии развивается развернутая морфологическая картина цирроза печени.
Симптомы первичного билиарного цирроза печени
Течение первичного билиарного цирроза печени может быть бессимптомным, медленным и быстропрогрессирующим. При бессимптомном течении заболевание обнаруживается на основании изменений лабораторных показателей – повышения активности щелочной фосфатазы, увеличения уровня холестерина, выявления АМА.
Наиболее типичным клиническим проявлением первичного билиарного цирроза печени служит кожный зуд, который предшествует появлению желтушного окрашивания склер и кожи. Кожный зуд может беспокоить в течение нескольких месяцев или лет, поэтому часто пациенты все это время безуспешно лечатся у дерматолога. Беспокоящий зуд приводит к множественным расчесам кожи спины, рук и ног. Желтуха обычно развивается спустя 6 месяцев-1,5 года после начала кожного зуда. У пациентов с первичным билиарным циррозом отмечаются боли в правом подреберье, гепатомегалия (селезенка чаще всего не увеличена).
В развернутой стадии первичного билиарного цирроза печени появляется субфебрилитет, нарастает желтуха, ухудшение самочувствия, истощение. Прогрессирующий холестаз вызывает диспепсические расстройства – диарею, стеаторею. Осложнениями первичного билиарного цирроза печени могут служить желчнокаменная болезнь, язвы 12-перстной кишки, холангиокарциномы.
В поздней стадии развивается остеопороз, остеомаляция, патологические переломы, геморрагический синдром, варикозное расширение вен пищевода. Гибель пациентов наступает от печеночноклеточной недостаточности, которая может провоцироваться портальной гипертензией, желудочно-кишечными кровотечениями, асицтом.
Диагностика первичного билиарного цирроза печени
Ранними диагностическими критериями первичного билиарного цирроза печени служат изменения биохимических показателей крови. При исследовании печеночных проб отмечается повышение активности щелочной фосфатазы, уровня билирубина, аминотрансфераз, рост концентрации желчных кислот. Характерно увеличение содержания меди и снижение уровня железа в сыворотке крови. Уже на ранних стадиях определяется гиперлипидемия – увеличение уровня холестерина, фосфолипидов, b-липопротеидов. Определяющее значение имеет выявление титра антимитохондриальных антител выше 1:40, повышение уровня IgM и IgG.
По данным УЗИ печени и МРТ печени внепеченочные желчные протоки не изменены. Для подтверждения первичного билиарного цирроза показано проведение биопсии печени с морфологическим исследованием биоптата.
Первичный билиарный цирроз печени дифференцируют от заболеваний, протекающих с обструкцией гепетобилиарного тракта и холестазом: стриктурами, опухолями печени, конкрементами, склерозирующим холангитом, аутоиммунным гепатитом, карциномой внутрипеченочных протоков, хроническим вирусным гепатитом С и др. В ряде случаев с целью дифференциальной диагностики прибегают к проведению ультрасонографии желчных путей, гепатобилисцинтиграфии, чрескожной чреспеченочной холангиографии, ретроградной холангиографии.
Лечение первичного билиарного цирроза печени
Терапия первичного билиарного цирроза печени включает назначение иммуносупрессивных, противовоспалительных, антифибротических препаратов, желчных кислот. Диета при первичном билиарном циррозе печени требует достаточного употребления белка, поддержания необходимой калорийности пищи, ограничения жиров.
К препаратам патогенетической терапии относятся глюкокортикостероиды (будесонид), цитостатики (метотрексат), колхицин, циклоспорин А, урсодезоксихолевая кислота. Длительный и комплексный прием препаратов позволяет улучшить биохимические показатели крови, замедлить прогрессирование морфологических изменений, развитие портальной гипертензии и цирроза.
Симптоматическая терапия первичного билиарного цирроза печени включает мероприятия, направленные на уменьшение кожного зуда (УФО, седативные препараты), потери костной массы (прим витамина D, препаратов кальция) и др. При рефрактерных к основной терапии формах первичного билиарного цирроза показана как можно более ранняя трансплантация печени.
Прогноз первичного билиарного цирроза печени
При бессимптомном течении первичного билиарного цирроза печени продолжительность жизни составляет 15-20 и более лет. Прогноз у пациентов с клиническими проявлениями значительно хуже – гибель от печеночной недостаточности наступает примерно в течение 7-8 лет. Значительно отягощает течение первичного билиарного цирроза печени развитие асцита, варикозного расширения вен пищевода, остеомаляции, геморрагического синдрома.
После трансплантации печени вероятность рецидива первичного билиарного цирроза достигает 15-30%.
*Импакт фактор за 2018 г. по данным РИНЦ
Журнал входит в Перечень рецензируемых научных изданий ВАК.
Читайте в новом номере
Первичный билиарный цирроз (ПБЦ) – это медленно прогрессирующее аутоиммунное заболевание печени, встречающиеся преимущественно у женщин. Наиболее часто билиарный цирроз развивается в возрасте от 40 до 50 лет и крайне редко – у людей моложе 25 лет. При гистологическом исследовании отмечаются воспалительные изменения портальных трактов и аутоиммунное разрушение внутрипеченочных желчных протоков. Это приводит к нарушению выделения желчи и задержке токсических веществ в печени, что является причиной снижения функции печени, фиброза, цирроза и печеночной недостаточности.
Литература
1. Gershwin ME, Mackay IR, Sturgess A, Coppel RL. Identification and specificity of a cDNA encoding the 70 kd mitochondrial antigen recognized in primary biliary cirrhosis. J Immunol 1987;138:3525–31.
2. Kaplan MM. Primary biliary cirrhosis. N Engl J Med 1996;335:1570–80.
3. Pares A, Rodes J. Natural history of primary biliary cirrhosis. Clin Liver Dis 2003;7:779–94.
4. Prince MI, Chetwynd A, Craig WL, Metcalf JV, James OF. Asymptomatic primary biliary cirrhosis: clinical features, prognosis, and symptom progression in a large population based cohort. Gut 2004;53:865–70. [Erratum, Gut 2004;53:1216.]
5. Bergasa NV. Pruritus and fatigue in primary biliary cirrhosis. Clin Liver Dis 2003;7: 879–900.
6. Prince M, Chetwynd A, Newman W, Metcalf JV, James OFW. Survival and symptom progression in a large geographically based cohort of patients with primary biliary cirrhosis: follow–up for up to 28 years. Gastroenterology 2002;123:1044–51.
7. Milkiewicz P, Heathcote EJ. Fatigue in chronic cholestasis. Gut 2004;53:475–7.
8. Forton DM, Patel N, Prince M, et al. Fatigue and primary biliary cirrhosis: association of globus pallidus magnetisation transfer ratio measurements with fatigue severity and blood manganese levels. Gut 2004;53: 587–92.
9. Poupon RE, Chretien Y, Chazouilleres O, Poupon R, Chwalow J. Quality of life in patients with primary biliary cirrhosis. Hepatology 2004;40:489–94.
10. Talwalkar JA, Souto E, Jorgensen RA, Lindor KD. Natural history of pruritus in primary biliary cirrhosis. Clin Gastroenterol Hepatol 2003;1:297–302.
11. Laurin JM, DeSotel CK, Jorgensen RA, Dickson ER, Lindor KD. The natural history of abdominal pain associated with primary biliary cirrhosis. Am J Gastroenterol 1994; 89:1840–3.
12. Watt FE, James OF, Jones DE. Patterns of autoimmunity in primary biliary cirrhosis patients and their families: a populationbased cohort study. QJM 2004;97:397–406.
13. Nakanuma Y. Are esophagogastric varices a late manifestation in primary biliary cirrhosis? J Gastroenterol 2003;38:1110–2.
14. Nijhawan PK, Thernau TM, Dickson ER, Boynton J, Lindor KD. Incidence of cancer in primary biliary cirrhosis: the Mayo experience. Hepatology 1999;29:1396–8.
15. Prince MI, James OF. The epidemiology of primary biliary cirrhosis. Clin Liver Dis 2003;7:795–819.
16. Poupon RE, Lindor KD, Pares A, Chazouilleres O, Poupon R, Heathcote EJ. Combined analysis of the effect of treatment with ursodeoxycholic acid on histologic progression in primary biliary cirrhosis. J Hepatol 2003;39:12–6.
17. Poupon R. Trials in primary biliary cirrhosis: need for the right drugs at the right time. Hepatology 2004;39:900–2.
18. Lee YM, Kaplan MM. Efficacy of colchicine in patients with primary biliary cirrhosis poorly responsive to ursodiol and methotrexate. Am J Gastroenterol 2003;98:205–8.
19. Kaplan MM, DeLellis RA, Wolfe HJ. Sustained biochemical and histologic remission of primary biliary cirrhosis in response to medical treatment. Ann Intern Med 1997; 126:682–8.
20. Bonis PAL, Kaplan M. Methotrexate improves biochemical tests in patients with primary biliary cirrhosis who respond incompletely to ursodiol. Gastroenterology 1999;117:395–9.
21. Leuschner M, Dietrich CF, You T, et al. Characterisation of patients with primary biliary cirrhosis responding to long term ursodeoxycholic acid treatment. Gut 2000;46: 121–6.
22. Locke GR III, Therneau TM, Ludwig J, Dickson ER, Lindor KD. Time course of histological progression in primary biliary cirrhosis. Hepatology 1996;23:52–6.
23. Corpechot C, Carrat F, Bahr A, Chretien Y, Poupon R–E, Poupon R. The effect of ursodeoxycholic acid therapy on the natural course of primary biliary cirrhosis. Gastroenterology 2005;128:297–303.
24. Degott C, Zafrani ES, Callard P, Balkau B, Poupon RE, Poupon R. Histopathological study of primary biliary cirrhosis and the effect of ursodeoxycholic acid treatment on histology progression. Hepatology 1999; 29:1007–12.
25. Therneau TM, Grambsch PM. Modeling survival data: extending the Cox model. New York: Springer, 2000:261–87.
26. Heathcote EJ, Cauch–Dudek K, Walker V, et al. The Canadian multicenter doubleblind randomized controlled trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology 1994;19:1149–56.
27. Poupon RE, Lindor KD, Cauch–Dudek K, Dickson ER, Poupon R, Heathcote EJ. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology 1997;113: 884–90.
28. Springer J, Cauch–Dudek K, O’Rourke K, Wanless I, Heathcote EJ. Asymptomatic primary biliary cirrhosis: a study of its natural history and prognosis. Am J Gastroenterol 1999;94:47–53.
29. Corpechot C, Carrat F, Bonnand AM, Poupon RE, Poupon R. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology 2000;32:1196–9.
30. Van Norstrand MD, Malinchoc M, Lindor KD, et al. Quantitative measurement of autoantibodies to recombinant mitochondrial antigens in patients with primary biliary cirrhosis: relationship of levels of autoantibodies to disease progression. Hepatology 1997;25:6–11.
31. Parikh–Patel A, Gold EB, Worman H, Krivy KE, Gershwin ME. Risk factors for primary biliary cirrhosis in a cohort of patients from the United States. Hepatology 2001; 33:16–21.
32. Howel D, Fischbacher CM, Bhopal RS, Gray J, Metcalf JV, James OF. An exploratory population–based case–control study of primary biliary cirrhosis. Hepatology 2000;31: 1055–60.
33. Sood S, Gow PJ, Christie JM, Angus PW. Epidemiology of primary biliary cirrhosis in Victoria, Australia: high prevalence in migrant populations. Gastroenterology 2004; 127:470–5.
34. Bittencourt PL, Farias AQ, Abrantes– Lemos CP, et al. Prevalence of immune disturbances and chronic liver disease in family members of patients with primary biliary cirrhosis. J Gastroenterol Hepatol 2004;19: 873–8.
35. Selmi C, Mayo MJ, Bach N, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology 2004;127:485– 92.
36. Invernizzi P, Battezzati PM, Crosignani A, et al. Peculiar HLA polymorphisms in Italian patients with primary biliary cirrhosis. J Hepatol 2003;38:401–6.
37. Jones DE, Donaldson PT. Genetic factors in the pathogenesis of primary biliary cirrhosis. Clin Liver Dis 2003;7:841–64.
38. Springer JE, Cole DE, Rubin LA, et al. Vitamin D–receptor genotypes as independent genetic predictors of decreased bone mineral density in primary biliary cirrhosis. Gastroenterology 2000;118:145–51.
39. Tanaka A, Lindor K, Gish R, et al. Fetal microchimerism alone does not contribute to the induction of primary biliary cirrhosis. Hepatology 1999;30:833–8.
40. Invernizzi P, Miozzo M, Battezatti PM, et al. The frequency of monosomy X in women with primary biliary cirrhosis. Lancet 2004;363:533–5.
41. Selmi C, Gershwin EM. Bacteria and human autoimmunity: the case of primary biliary cirrhosis. Curr Opin Rheumatol 2004; 16:406–10.
42. Selmi C, Balkwill DL, Invernizzi P, et al. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic–metabolizing bacterium. Hepatology 2003;38:1250–7.
43. Xu L, Shen Z, Guo L, et al. Does a betavirus infection trigger primary biliary cirrhosis? Proc Natl Acad Sci U S A 2003;100: 8454–9.
44. Selmi C, Ross SA, Ansari A, et al. Lack of immunological or molecular evidence for a role of mouse mammary tumor retrovirus in primary biliary cirrhosis. Gastroenterology 2004;127:493–501.
45. Long SA, Quan C, Van de Water J, et al. Immunoreactivity of organic mimeotopes of the E2 component of pyruvate dehydrogenase: connecting xenobiotics with primary biliary cirrhosis. J Immunol 2001;167:2956– 63.
46. Leung PS, Quan C, Park O, et al. Immunization with a xenobiotic 6–bromohexane bovine serum albumin conjugate induces antimitochondrial antibodies. J Immunol 2003;170:5326–32.
47. Bruggraber SF, Leung PS, Amano K, et al. Autoreactivity to lipoate and a conjugated form of lipoate in primary biliary cirrhosis. Gastroenterology 2003;125:1705–13.
48. Gershwin ME, Ansari AA, Mackay IR, et al. Primary biliary cirrhosis: an orchestrated immune response against epithelial cells. Immunol Rev 2000;174:210–25.
49. Kita H, Matsumura S, He X–S, et al. Quantitative and functional analysis of PDC–E2 specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest 2002;109:1231–40.
50. Kita H, Naidenko OV, Kronenberg M, et al. Quantitation and phenotypic analysis of natural killer T cells in primary biliary cirrhosis using a human CD1d tetramer. Gastroenterology 2002;123:1031–43.
51. Shimoda S, Van de Water J, Ansari A, et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest 1998;102:1831– 40.
52. Odin JA, Huebert RC, Casciola–Rosen L, LaRusso NF, Rosen A. Bcl–2–dependent oxidation of pyruvate dehydrogenase–E2, a primary biliary cirrhosis autoantigen, during apoptosis. J Clin Invest 2001;108:223–32.
53. Amano K, Leung PS, Xu Q, et al. Xenobiotic– induced loss of tolerance in rabbits to the mitochondrial autoantigen of primary biliary cirrhosis is reversible. J Immunol 2004;172:6444–52.
54. Matsumura S, Kita H, He XS, et al. Comprehensive mapping of HLA–A0201–restricted CD8 T–cell epitopes on PDC–E2 in primary biliary cirrhosis. Hepatology 2002;36: 1125–34.
55. Worman HJ, Courvalin JC. Antinuclear antibodies specific for primary biliary cirrhosis. Autoimmun Rev 2003;2:211–7.
56. Ghent CN, Carruthers SG. Treatment of pruritus in primary biliary cirrhosis with rifampin: results of a double–blind, crossover, randomized trial. Gastroenterology 1988; 94:488–93.
57. Cohen LB, Ambinder EP, Wolke AM, Field SP, Schaffner F. Role of plasmapheresis in primary biliary cirrhosis. Gut 1985;26: 291–4.
58. Levy C, Lindor KD. Management of osteoporosis, fat–soluble vitamin deficiencies, and hyperlipidemia in primary biliary cirrhosis. Clin Liver Dis 2003;7:901–10.
59. Ormarsdottir S, Ljunggren O, Mallmin H, Olsson R, Prytz H, Loof L. Longitudinal bone loss in postmenopausal women with primary biliary cirrhosis and well–preserved liver function. J Intern Med 2002;252:537–41.
60. Boulton–Jones JR, Fenn RM, West J, Logan RF, Ryder SD. Fracture risk of women with primary biliary cirrhosis: no increase compared with general population controls. Aliment Pharmacol Ther 2004;20:551–7.
61. Guanabens N, Pares A, Ros I, et al. Alendronate is more effective than etidronate for increasing bone mass in osteopenic patients with primary biliary cirrhosis. Am J Gastroenterol 2003;98:2268–74.
62. Menon KV, Angulo P, Boe GM, Lindor KD. Safety and efficacy of estrogen therapy in preventing bone loss in primary biliary cirrhosis. Am J Gastroenterol 2003;98:889–92.
63. Longo M, Crosignani A, Battezzati PM, et al. Hyperlipidaemic state and cardiovascular risk in primary biliary cirrhosis. Gut 2002;51:265–9.
64. Thornton JR, Triger DR. Variceal bleeding is associated with reduced risk of severe cholestasis in primary biliary cirrhosis. Q J Med 1989;71:467–71.
65. Boyer TD, Kokenes DD, Hertzler G, Kutner MH, Henderson JM. Effect of distal splenorenal shunt on survival of patients with primary biliary cirrhosis. Hepatology 1994;20:1482–6.
66. Pares A, Caballeria L, Rodes J, et al. Long–term effects of ursodeoxycholic acid in primary biliary cirrhosis: results of a double–blind controlled multicentric trial. J Hepatol 2000;32:561–6.
67. Poupon RE, Bonnand AM, Chretien Y, Poupon R. Ten–year survival in ursodeoxycholic acid–treated patients with primary biliary cirrhosis. Hepatology 1999;29:1668–71.
68. Lindor KD, Jorgenson RA, Therneau TM, Malinchoc M, Dickson ER. Ursodeoxycholic acid delays the onset of esophageal varices in primary biliary cirrhosis. Mayo Clin Proc 1997;72:1137–40.
69. Goulis J, Leandro G, Burroughs AK. Randomised controlled trials of ursodeoxycholic– acid therapy for primary biliary cirrhosis: a meta–analysis. Lancet 1999;354: 1053–60.
70. Gluud C, Christensen E. Ursodeoxycholic acid for primary biliary cirrhosis. Cochrane Database Syst Rev 2002;1: CD000551.
71. Corpechot C, Carrat F, Bahr A, Poupon RE, Poupon R. The impact of ursodeoxycholic (UDCA) therapy with or without liver transplantation (OLT) on long–term survival in primary biliary cirrhosis. Hepatology 2003;34:519A. abstract.
72. Kaplan MM, Alling DW, Zimmerman HJ, et al. A prospective trial of colchicine for primary biliary cirrhosis. N Engl J Med 1986; 315:1448–54.
73. Vuoristo M, Farkkila M, Karvonen AL, et al. A placebo–controlled trial of primary biliary cirrhosis treatment with colchicine and ursodeoxycholic acid. Gastroenterology 1995;108:1470–8.
74. Kaplan MM, Schmid C, Provenzale D, Sharma A, Dickstein G, McKusick A. A prospective trial of colchicine and methotrexate in the treatment of primary biliary cirrhosis. Gastroenterology 1999;117:1173–80.
75. Kaplan MM, Cheng S, Price LL, Bonis PA. A randomized controlled trial of colchicine plus ursodiol versus methotrexate plus ursodiol in primary biliary cirrhosis: tenyear results. Hepatology 2004;39:915–23.
76. Almasio P, Floreani A, Chiaramonte M, et al. Multicentre randomized placebo–controlled trial of ursodeoxycholic acid with or without colchicine in symptomatic primary biliary cirrhosis. Aliment Pharmacol Ther 2000;14:1645–52.
77. Vela S, Agrawal D, Khurana S, Singh P. Colchicine for primary biliary cirrhosis: a meta–analysis of prospective controlled trials. Gastroenterology 2004;126:A671– A672. abstract.
78. Cronstein BN, Naime D, Ostad E. The antiinflammatory mechanism of methotrexate: increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J Clin Invest 1993;92:2675–82.
79. Hendrickse M, Rigney E, Giaffer MH, et al. Low–dose methotrexate is ineffective in primary biliary cirrhosis: long–term results of a placebo–controlled trial. Gastroenterology 1999;117:400–7.
80. Gonzalez–Koch A, Brahm J, Antezana C, Smok G, Cumsille MA. The combination of ursodeoxycholic acid and methotrexate for primary biliary cirrhosis is not better then ursodeoxycholic acid alone. J Hepatol 1997;27:143–9.
81. Combes B, Emerson SS, Flye NL. The primary biliary cirrhosis (PBC) ursodiol (UDCA) plus methotrexate (MTX) or its placebo study (PUMPS) — a multicenter randomized trial. Hepatology 2003;38:210A. abstract.
82. Leuschner M, Maier KP, Schlictling J, et al. Oral budesonide and ursodeoxycholic acid for treatment of primary biliary cirrhosis: results of a prospective double–blind trial. Gastroenterology 1999;117:918–25.
83. Rautiainen H, Karkkainen P, Karvonen A–L, et al. Budesonide combined with UDCA to improve liver histology in primary biliary cirrhosis: a three–year randomized trial. Hepatology 2005;41:747–52.
84. Angulo P, Jorgensen RA, Keach JC, Dickson ER, Smith C, Lindor KD. Oral budesonide in the treatment of patients with primary biliary cirrhosis with a suboptimal response to ursodeoxycholic acid. Hepatology 2000;31:318–23.
85. Mitchison HC, Palmer JM, Bassendine MF, Watson AJ, Record CO, James OF. A controlled trial of prednisolone treatment in primary biliary cirrhosis: three–year results. J Hepatol 1992;15:336–44.
86. Angulo P, Patel T, Jorgensen RA, Therneau TM, Lindor KD. Silymarin in the treatment of patients with primary biliary cirrhosis with a suboptimal response to ursodeoxycholic acid. Hepatology 2000;32: 897–900.
87. Nakai S, Masaki T, Kurokohchi K, Deguchi A, Nishioka M. Combination therapy of bezafibrate and ursodeoxycholic acid in primary biliary cirrhosis: a preliminary study. Am J Gastroenterol 2000;95:326–7.
88. Reddy A, Prince M, James OF, Jain S, Bassendine MF. Tamoxiphen: a novel treatment for primary biliary cirrhosis? Liver Int 2004;24:194–7.
89. Leuschner M, Holtmeier J, Ackermann H, Leuschner U. The influence of sulindac on patients with primary biliary cirrhosis that responds incompletely to ursodeoxycholic acid: a pilot study. Eur J Gastroenterol Hepatol 2002;14:1369–76.
90. Hoofnagle JH, Davis GL, Schafer DF, et al. Randomized trial of chlorambucil for primary biliary cirrhosis. Gastroenterology 1986;91:1327–34.
91. James OF. D–penicillamine for primary biliary cirrhosis. Gut 1985;26:109–13.
92. Christensen E, Neuberger J, Crowe J, et al. Beneficial effect of azathioprine and prediction of prognosis in primary biliary cirrhosis: final results of an international trial. Gastroenterology 1985;89:1084–91.
93. Lombard M, Portmann B, Neuberger J, et al. Cyclosporin A treatment in primary biliary cirrhosis: results of a long–term placebo controlled trial. Gastroenterology 1993;104: 519–26.
94. The results of a randomized double blind controlled trial evaluating malotilate in primary biliary cirrhosis. J Hepatol 1993; 17:227–35.
95. McCormick PA, Scott F, Epstein O, Burroughs AK, Scheuer PJ, McIntyre N. Thalidomide as therapy for primary biliary cirrhosis: a double–blind placebo controlled pilot study. J Hepatol 1994;21:496–9.
96. Talwalkar JA, Angulo P, Keach J, Petz JL, Jorgensen RA, Lindor KD. Mycophenolate mofetil for the treatment of primary biliary cirrhosis in patients with an incomplete response to ursodeoxycholic acid. J Clin Gastroenterol 2005;39:168–71.
97. MacQuillan GC, Neuberger J. Liver transplantation for primary biliary cirrhosis. Clin Liver Dis 2003;7:941–56.
98. Neuberger J. Liver transplantation for primary biliary cirrhosis: indications and risk of recurrence. J Hepatol 2003;39: 142–8.
Читайте также:
 Пожалуйста, не занимайтесь самолечением!При симпотмах заболевания - обратитесь к врачу.
Пожалуйста, не занимайтесь самолечением!При симпотмах заболевания - обратитесь к врачу.
Copyright © Иммунитет и инфекции