
Мнс и предрасположенность к инфекциям аутоиммунной патологии

В наше время заболевания щитовидной железы являются актуальной проблемой. Так, по данным ВОЗ (всемирная организация здравоохранения), около 31% населения планеты страдает от тиреоидных заболеваний, причем в России заболевания такого характера встречаются довольно часто и составляют около 40% населения, а в некоторых районах доходит и до 95%. Самыми распространенными являются аутоиммунные заболевания щитовидной железы, а именно аутоиммунный тиреоидит или (тиреоидит Хашимото) и диффузный токсический зоб или (болезнь Грейвса), которые составляют 10-15%. Аутоиммунные заболевания обусловлены тем, что иммунная система человека начинает уничтожать клетки щитовидной железы, вследствие чего железа перестает выполнять свои определенные функции. Обычно, такие заболевания чаще встречаются у женщин, около 3-4%, но выявлены случаи и у мужчин, 1-1,5. Также данная патология наблюдается после прохождения курса лечения тиреоидными препаратами, а также у женщин во время беременности. На сегодняшний день выявлено три главных фактора, позволяющих выявить аутоиммунные заболевания, а именно: повышение уровня специфических аутоантител к рецептору ТТГ, тиреопероксидазе и тиреоглобулину, изменение концентрации ТТГ и изменение концентрации Т3, Т4 в крови. Вышеуказанные факторы могут вызываться генетическими, стресс-ассоциированными причинами и ненормированным приемом препаратов. Для лучшего понимания необходимо рассмотреть механизм влияния вышеприведенных причин на дисфункцию щитовидной железы.
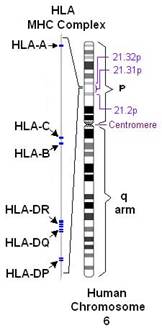
Генетические нарушения являются первыми причинами появления аутоиммунных заболеваний. К ним могут относиться различные явления, например: наличие генов предрасположенности, структурные и функциональные нарушения в гене, кодирующим определенные элементы и вещества щитовидной железы. Среди общеизвестных генов предрасположенности наиболее значимый риск создают HLA-DR β1-Arg74 (человеческий лейкоцитарный антиген сублокуса DR, с аргинином в позиции 74 β-цепи), относятся к Т-клеточными эпитопами. Также, что является важным, гены главного комплекса гистосовместимости (МНС) класса II обеспечивают предрасположенность к аутоиммунным заболеваниям. Тем не менее было подтверждено, что в домене МНС присутствуют различные гены предрасположенности, которые также участвуют в развитии заболеваний щитовидной железы. Некоторые гены уникальны для диффузного токсического зоба, такие как HLA-DR3 или для тиреоидита, HLA-DR5, которые находятся в q-плече 6 хромосомы человека. (рис. 1(а)). К факторам окружающей среды, являющимися триггерными в развитии болезни Грейвса и тиреоидита, относят: несбалансированная диета, избыточное поступление йода в организм, а также курение. [4, с.74-75]
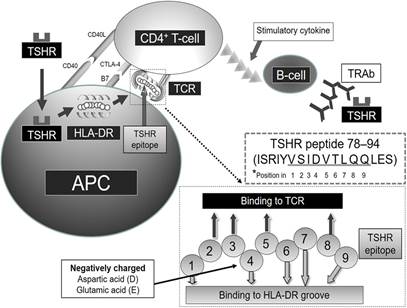
Также важным является антитела к тиреоглобулину (TgAb), которые имеют схожий патогенез. Они представляют собой иммуноглобулин класса G. С клинической точки зрения, определение наличие TgAb в крови является важным так как они мешают прогнозированию дисфункции щитовидной железы. Повышение тиреоглобулина может свидетельствовать о разных заболеваниях, например рак щитовидной железы, но наличие аутоантител затрудняет достоверно определить тиреоглобулин в крови, что может повлиять на своевременное обнаружения заболевания. При уменьшении количества тиреоглобулина так же влияет на функцию щитовидной железы, как и уменьшение пероксидазы. [9] .Интересный факт, что гены предрасположенности HLA-DR3-5, являются главными антигенами тиреоглобулина и вызывают те же нарушения что и в TSHR, поэтому при болезни Хашимото наблюдается повышение антител, как и к TSHR, так и к ТРО.
IL-34 был впервые описан в 2008 году, и только недавно было выявлено его участие в развитии многих аутоиммунных заболеваний, однако неясно, является ли IL-34 регуляторным фактором при аутоиммунном тиреоидите. Он экспрессируется на эпителиальных клетках щитовидной железы и участвует в апоптозной резистентности тироцитов при тиреоидите и может быть потенциальным индикатором для оценки повреждения тироцитов. Но при аутоиммунных заболеваниях его экспрессия значительно снижается в ткани щитовидной железы, из-за чего может возникать массовая гибель тироцитов[7]
Ко второй группе причин относиться тиреоидит во время беременности и послеродовой, и терапия лекарственными средствами. Дисфункция щитовидной железы во время беременности классифицируется как форма гипотиреоза (положительность аутоантител щитовидной железы), гипертиреоза, аутоиммунное заболевание и также узлы щитовидной железы, рак и йодная недостаточность.[2] Эти состояния могут оказывать неблагоприятное воздействие на мать и плод, включая потерю беременности, преждевременные роды, недоношенность плода, отслойку плаценты и послеродовое кровоизлияние. Существует доказательство того, что аутоиммунитет щитовидной железы при ее нарушении отрицательно влияет на зачатие и результаты беременности, так как антитела TSHR преимущественно относятся к иммуноглобулинам класса IgG, которые могут проникают через плацентарный барьер и агрессивно влиять на развитие плода, а также оказывать негативное влияние на щитовидную железу и повышать риск врожденных отклонений. Необходимо подчеркнуть, что гормоны щитовидной железы оказывают влияние на развитие нервной и половой систем плода, органогенез и созревание гипоталамуса, гипофиза и щитовидной железы, так, в качестве примера, приведен случай, связанный с возникновением у ребенка 2.5-летняя болезни Грейвса. Девочка была госпитализирована с тахикардией и субфебрильной температурой, общей слабостью. При физическом обследовании у ребенка была обнаружена увеличенная щитовидная железа, а при лабораторных исследованиях выявили увеличение как FT3 (> 30 пг / мл), так и FT4 (3,43 нг / дл), а уровни антител против тиреоглобулина и к пероксидазе не были идентифицированы. Была диагностирована болезнь Грейвса. Через 2 месяца после поставленного диагноза у матери ребенка была диагностирована болезнь Хашимото. [8]
Читайте также:
|
| Патоген | Человеческие белки | Длина совпадающих первичных последовательностей |
| Человеческий цитомегаловирус | Молекулы HLA-DR | 5 аминокислот |
| Вирус полиомиелита | рецептор к ацетилхолину | 6 аминокислот |
| Вирус папилломы Е2 | рецептор к инсулину | 6 аминокислот |
| Гликопротеин вируса бешенства | рецептор к инсулину | 6 аминокислот |
| Klebsiella pneumonia nitrogenase | молекула HLA – В27 | 6 аминокислот |
| Аденовирус 12 Е1В | a–глиадин | 7 аминокислот |
| ВИЧ, р24 | постоянный домен IgG | 8 аминокислот |
| Вирус кори, Р3 | кортикотропин | 7 аминокислот |
| Вирус кори, Р3 | основной белок миелина | 7 аминокислот |
Считается, что молекулярная мимикрия участвует в индукции ревматизма: Streptococcus pyogenes группы А (β-гемолитический стрептококк группы А) вызывает синтез антител, которые реагируют с белком М стрептококка и с сердечной мышцей. Еще одним ярким примером является возникновение аутоиммунного поствакцинального энцефалита у некоторых людей после проведения им вакцинации против бешенства. Так как один из пептидов основного белка миелина имеет участок, высоко гомологичный пептиду Р3 вируса кори, то одним из редких осложнений вакцинации против кори является аутоиммунное поражение миелиновой оболочки, которое может послужить пусковым механизмом возникновения рассеянного склероза.
Компьютерный анализ показал наличие гомологичных последовательностей между основным белком миелина и большим числом пептидов, полученных из вирусов человека и животных, включая вирусы гриппа, полиомы, аденовирусы, вирусы полиомиелита, Эпштейна-Барра и гапатита В. Особенно высокая степень гомологии обнаружена между белком одним из ферментов вируса гепатита В и основным белком миелина. Кроликов иммунизировали эти пептидом из вируса гепатита В, и в результате у них был обнаружен синтез антител и пролиферация Т-лимфоцитов, которые перекрестно реагировали с основным белком миелина.
Эти данные показывают, что инфицирование определенными вирусами, экспрессирующими эпитопы, обладающие молекулярной мимикрией к собственным секвестрированным антигенам, таким как основной белок миелина, может индуцировать аутоиммунный ответ к этим компонентам. Чувствительность к такому типу аутоиммунных реакций может также зависеть от МНС-гаплотипа индивидуума, так как определенные молекулы МНС I и II класса могут более эффективно представлять гомологичные пептиды патогенов, вызывая активацию аутореактивных Т-лимфоцитов.
Другим механизмом, способствующим индукции аутоиммунных процессов при инфекции, в первую очередь вирусной, является неадекватная экспрессия молекул МНС I класса на инфицированных клетках, что может привести к активации аутореактивных клонов Т-лимфоцитов. Показано, что у здоровых людей на β-клетках поджелудочной железы белки МНС I класса почти не экспрессируются, в то время как у больных инсулинозависимым диабетом обнаружен высокий уровень экспрессии этих белков. Установлено также, что вирусная инфекция может вызвать локальный воспалительный ответ в пораженном органе, приводящий к повышению продукции IFN-γ, который в свою очередь стимулирует экспрессию молекул МНС на β-клетках поджелудочной железы.
У некоторых людей иммунный ответ против инфекции заканчивается после элиминации патогена, вслед за этим заканчивается и возникший в результате эпитопного распространения ответ против собственных антигенов инфицированной ткани (аутоиммунный компонент инфекционного заболевания). У других же людей (генетически предрасположенных, например) даже после окончания иммунного ответа против элиминированного патогена аутоиммунный ответ против собственных тканей продолжается и прогрессирует. У них аутоиммунный компонент инфекционного заболевания перерастает в истинное аутоиммунное заболевание, продолжающееся и после удаления триггерного фактора.
Эпитопное распространение, по-видимому, лежит в основе патогенеза гемолитической анемии, возникшей при инфицировании некоторых людей бактерией Mycoplasma pulmonis. Антитела против этого патогена, относящееся к IgM, могут присоединяться как к собственным полисахаридам бактерий, так и к структурно похожим олигосахаридам антигена I эритроцитов, вызывая их комплементзависимый лизис. Таким образом происходит эпитопное распространение антительного ответа с бактериального компонента на структурно похожий компонент собственных клеток организма.
Механизм эпитопного распространения, скорее всего, участвует в патогенезе экспериментального аллергического энцефаломиелита (ЭАЭ) у мышей, вызванного вирусом мышиного энцефаломиелита Тейлера (Theiler), который является природным патогеном для мышей. Повреждения, вызванные вирусом у этих мышей, приводят в результате эпитопного распространения к активации клонов, аутореактивных к протеолипидам миелиновой оболочки. Именно эти клоны активируются при индукции ЭАЭ в результате иммунизации мышей миелиновыми белками или пептидами в результате эпитопного распространения без участия вируса.
Ещё одним механизмом, способствующим возникновению аутоиммунных заболеваний при инфекциях, является поликлональная активация лимфоцитов.
Причиной такой активации могут служить суперантигены – белки, продуцируемые бактериями или вирусами, которые, присоединяясь к цепям антигенраспознающего рецептора (неспецифически) и одновременно к молекулам МНС, представляющим пептиды суперантигена на поверхности антигенпредставляющих клеток, вызывают сильную поликлональную активацию Т-лимфоцитов, в том числе и аутореактивных, тем самым индуцируя аутоиммунный ответ. Примером таких суперантигенов может служить суперантиген Mycoplasma arthritidis, вызывающий артрит.
Поликлональную активацию В-лимфоцитов могут вызвать самые различные вирусы и бактерии: многие граммотрицательные бактерии, вирус Эпштейна-Барра и цитомегаловирусы, которые являются известными поликлональными активаторами, индуцирующими пролиферацию многочисленных клонов В-лимфоцитов, которые синтезируют IgM в отсутствие Т-хелперов. Если при этом активируются и аутореактивные клоны В-клеток, то начинается синтез аутоантител. Например, при инфекционном мононуклеозе, который вызывается вирусом Эпштейна-Барра, синтезируется множество аутоантител против различных собственных антигенов, включая антитела против Т- и В-лимфоцитов, антиядерные антитела и ревматоидный фактор. Подобным же образом лимфоциты больных СКВ продуцируют большие количества IgM против различных собственных антигенов. У многих больных СПИДом также обнаруживается высокий уровень аутоантител к эритроцитам и тромбоцитам. Эти больные часто бывают одновременно инфицированы и другими вирусами, в том числе вирусом Эпштейна-Барра и ЦМВ, которые также вызывают поликлональную В-клеточную активацию.
В последнее время интенсивно изучается ещё один механизм индукции аутоиммунной патологии, связанный с костимулирующей функцией антигенпредставляющих клеток (АПК), которая активируется инфекционными патогенами. Некоторые инфекционные агенты, паразитируя внутриклеточно в АПК, могут обеспечивать костимулирующие сигналыдля аутореактивных лимфоцитов, что может привести к их пролиферации, активации и нарушению периферических механизмов иммунологической толерантности к собственным антигенам. В роли таких АПК чаще всего выступают дендритные клетки (ДК).
Однако у некоторых людей абортивная активация может быть несовершенной, и активированные аутореактивные Т-лимфоциты атакуют собственные ткани организма. Такому срыву периферической толерантности может способствовать целый ряд факторов: генетическая предрасположенность, наличие вторичных иммунодефицитов и особенно – наличие очагов хронической инфекции.
Из изложенного видно, что среди неблагоприятных факторов внешней среды инфекции принадлежит ведущая роль в возникновении аутоиммунных заболеваний. Инфекционные агенты провоцируют нарушения механизмов периферической толерантности путем воздействия на различные звенья врожденного и адаптивного иммунитета, начиная с процессинга антигенов в АПК и заканчивая пролиферацией и дифференцировкой аутореактивных клонов эффекторных лимфоцитов. Исследования в этой области призваны углубить понимания механизмов патогенеза этой широко распространенной группы заболеваний человека, что позволит совершенствовать диагностику, лечение и разработать меры их профилактики.
Дата добавления: 2015-01-19 ; просмотров: 74 ; Нарушение авторских прав
АУТОИММУННЫЕ БОЛЕЗНИ И БОЛЕЗНИ С СИНДРОМАМИ ИММУННОГО ВОСПАЛЕНИЯ
Описание: Старение и некоторые заболевания приводят к тому что появляются антитела и Tлимфоциты направленные против собственных антигенов развиваются аутоиммунные реакции. Для диагностики аутоиммунных заболеваний применяют разнообразные исследования. В развитии аутоиммунных заболеваний играют роль наследственная предрасположенность неблагоприятное действие факторов окружающей среды нарушения иммунитета. Важную роль в развитии аутоиммунных заболеваний играют факторы окружающей среды например ультрафиолетовое излучение при СКВ и бактериальная.
Дата добавления: 2015-08-27
Размер файла: 25.99 KB
Работу скачали: 4 чел.
Поделитесь работой в социальных сетях
Если эта работа Вам не подошла внизу страницы есть список похожих работ. Так же Вы можете воспользоваться кнопкой поиск
АУТОИММУННЫЕ БОЛЕЗНИ И БОЛЕЗНИ С СИНДРОМАМИ ИММУННОГО ВОСПАЛЕНИЯ
В норме иммунный ответ развивается лишь на чужеродные или измененные собственные антигены. Старение и некоторые заболевания приводят к тому, что появляются антитела и T-лимфоциты, направленные против собственных антигенов, развиваются аутоиммунные реакции. Разнообразие клинических проявлений аутоиммунных заболеваний объясняется различиями в локализации, выраженности и механизмах повреждения собственных тканей и органов. Аутоиммунные болезни это болезни, в патогенезе которых лимфоциты распознают нативные молекулы мембран собственных клеток или межклеточного вещества и инициируют иммунное воспаление.
Аутоиммунное заболевание это заболевание, обусловленное аутоантителами (антителами к собственным антигенам) и цитотоксическими T-лимфоцитами, направленными против собственных антигенов. Четкую связь между развитием аутоиммунного заболевания и появлением аутоантител или цитотоксических T-лимфоцитов к собственным антигенам удается выявить не всегда. Для диагностики аутоиммунных заболеваний применяют разнообразные исследования.
Этиология и патогенез. В развитии аутоиммунных заболеваний играют роль наследственная предрасположенность, неблагоприятное действие факторов окружающей среды, нарушения иммунитета. Для многих аутоиммунных заболеваний выявлена связь с наследованием определенных генов HLA, генов иммуноглобулинов и антигенраспознающего рецептора T-лимфоцитов. Важную роль в развитии аутоиммунных заболеваний играют факторы окружающей среды, например ультрафиолетовое излучение при СКВ и бактериальная инфекция при реактивных артритах. Сочетание генетической предрасположенности с неблагоприятным действием факторов внешней среды, вероятно, стимулирует выработку цитокинов T-лимфоцитами, которые, в свою очередь, стимулируют пролиферацию и дифференцировку B-лимфоцитов и продукцию аутоантител. Развитие аутоиммунных реакций может быть обусловлено нарушением продукции антиидиотипических антител, контролирующих выраженность и продолжительность иммунного ответа. При многих аутоиммунных заболеваниях отмечается повышение активности тех клонов T-хелперов, которые стимулируют образование аутоантител. Показано, что лимфоциты CD8 (которые в норме выступают в роли T-супрессоров и цитотоксических T-лимфоцитов) при аутоиммунных заболеваниях могут стимулировать пролиферацию B-лимфоцитов и синтез антител. Некоторые из этих антител связываются с растворимыми антигенами и в виде иммунных комплексов откладываются в тканях, вызывая воспаление. Другие, непосредственно связываясь с тканевыми антигенами и комплементом, приводят к повреждению тканей. В качестве аутоантигенов могут выступать любые ткани, клетки и компоненты плазмы, в том числе сами иммуноглобулины. Так, ревматоидный фактор, например, это аутоантитела к IgG.
К аутоиммунным заболеваниям не относятся:
1. Патологические процессы, при которых имеется повреждение тканей иммунными механизмами. Иммунный ответ против патогенного антигена всегда сопровождается повреждением собственных тканей потому, что при проникновении патогена во внутреннюю среду он вступает в тесную связь с клетками и межклеточным веществом: разрушая патоген, иммунная система разрушает и окружающие собственные ткани .
Но это будут не аутоиммунные процессы, поскольку направлены против микробных антигенов и разрушение тканей наступает потому, что микробные продукты оказались тесно связанными с нашими тканями.
2. Денатурация собственных молекул организма химическими веществами (медикаменты, химические добавки к пище, факторы химических производственных процессов и другие вредности подобного рода) и превращение их в антигены. И в этих случаях альтерацию собственных тканей иммунными механизмами тоже неправильно называть аутоиммунным процессом, поскольку иммунная система борется с внешними повреждениями на поверхности собственных тканей.
Молекулы собственных клеток и межклеточного вещества не объект для распознавания неиммунных Т-лимфоцитами до тех пор, пока они не станут иммунными. Это происходит в результате какого-то предшествующего патологического процесса, приводящего к альтерации тканей и воспалению.
Иммунный Т-лимфоцит, отличается от неиммунного Т-лимфоцита:
1) для активации эффекторной функции иммунному лимфоциту достаточно только сигнала с его рецептора (т.е. он не зависит от костимуляторных взаимодействий);
Поэтому инфекции способны инициировать аутоиммунные процессы по следующим механизмам.
• Микробные суперантигены вызывают поликлональную активацию. Некоторые клоны лимфоцитов с реактивностью к своим антигенам могут войти в режим эффекторного иммунного ответа.
• Деструкция тканей патогеном (цитопатогенное действие вирусов, бактерий и др.) приводит к попаданию тканевых антигенов в активированные дендритные клетки, которые транспортируют все антигены в периферические лимфоидные органы, где есть условия для индукции продуктивного иммунного ответа.
• Индуцированное патогеном локальное доиммунное воспаление сопровождается выработкой провоспалительных цитокинов, которые способны индуцировать экспрессию на клетках тканей (не профессиональных антигенпредставляющих клеток) молекулы МНС со своими пептидами, что потенциально создает условия для инициации иммунного ответа на свои антигены.
Таким образом, начавшись в связи с инфекцией аутоиммунное воспаление не может нормальным образом остановиться, потому что аутоантиген не исчезает, пока вся ткань, его экспрессирующая, не будет разрушена и выброшена из организма .
К эффекторным механизмам нормального иммунного ответа относятся
• Антитела, комплемент, фагоцитоз, сосудистые и гладкомышечные реакции, опосредованные медиаторами тучных клеток и базофилов.
• ГЗТ: клетки-исполнители активированные макрофаги, клетки-инициаторы и регуляторы CD4+ Thl.
• Деструкция клеток-мишеней CD8+ ЦТЛ.
У разных пациентов аутоантигены-мишени одни и те же в пределах нозологии.
Таблица 1. Примеры аутоиммунных болезней человека
II тип повреждения тканей антитела к клеточным или матриксным антигенам
Аутоиммунная гемолитическая анемия
Разрушение эритроцитов комплементом и фагоцитозом - анемия
Интегрин тромбоцитов GpIIb/IIIa (рецептор для фибриногена)
Разрушение тромбоцитов кровоточивость
Неколлагеновый домен молекул коллагена IV типа базальных мембран

Аутоимунные тиреопатии (АТ) занимают ведущее место в структуре заболеваний щитовидной железы (ЩЗ) [2, 7]. К АТ относятся как заболевания, сопровождающиеся гипертиреозом – диффузный токсический зоб (ДТЗ), так и гипотиреозом – аутоиммунный тиреоидит Хашимото (АТХ) [2, 7, 23].
Наличием тесной взаимосвязи между иммунной и эндокринной системами обусловливаются многие особенности патогенеза АТ [6, 12, 21].
Рецепторы к тиреотропному гормону (ТТГ) обнаружены на многих клетках иммунной системы (моноцитах, макрофагах, дендритных клетках и Т-лимфоцитах). ТТГ может усиливать как Т-зависимую, так и Т-независимую продукцию антител. Наряду с центральной выработкой ТТГ, показана его периферическая продукция дендритными клетками, моноцитами и Т-лимфоцитами [8].
На клетках иммунной системы выявлена экспрессия ядерных рецепторов к трийодтиронину (Т3) (T3Rα1 и T3Rα2). В экспериментах на нокаутных по генам t3rα1 и t3rαα2 мышах отмечается выраженное падение количества иммунокомпетентных клеток в селезенке и дефекты пролиферации предшественников В-лимфоцитов [16].
У пациентов с тиреотоксикозом на фоне супрессивной терапии L-тироксином выявлен рост концентрации в сыворотке крови ИЛ-18, sИЛ-2R и увеличение количества NK-клеток. После отмены L-тироксина и перехода в гипотиреоидное состояние параметры клеточного иммунитета возвращались к норме [17].
На дендритных клетках щитовидной железы выявлена экспрессия рецепторов к Т3, что свидетельствует о возможности тиреоидных гормонов оказывать влияние на процессы пролиферации, дифференцировки и соответственно на функцию дендритных клеток. Физиологические концентрации Т3 вызывают экспрессию маркеров созревания дендритных клеток (МНС II класса, CD80, CD86 и CD40), усиливают продукцию ИЛ-12, стимулируют способность ДК вызывать пролиферацию и созревание наивных Т-клеток, продукцию ими ИФ-γ [22].
Значимый вклад в развитие аутоиммунного процесса при АТ вносит наследственная предрасположенность. Давно известно о наличии генов, которые влияют на формирование аутоиммунной патологии. По результатам исследования генома больных АТ выявлены локусы хромосом 2 (2q33), 6 (6p21), 8(8q24), 12 (12q22) и 13 (13q32), гены которых участвуют в развитии заболевания [5].
Наиболее важной является хромосома 2, на своем длинном плече она несет последовательность генов, которая отвечает за синтез ингибирующего рецептора CTLA-4 (cytotoxic T-lymphocyte antigen-4). CTLA-4 – это мощный ингибитор активации Т-лимфоцитов.
У людей, страдающих АТ, дефект именно в этом локусе провоцирует развитие иммунного процесса. При воздействии на щитовидную железу триггерных факторов (избыточное потребление йода, травмы ПЖ, инфекционные заболевания, вызванные вирусом гепатита C, HTLV-1, EBV, парвовирусом B19, Yersinia enterocolitica и др.) происходит повреждение тиреоцитов. Это запускает процесс миграции антигенпрезентирующих клеток (АПК) к месту повреждения и захват АПК аутоантигенов, с целью дальнейшей презентации аутоантигенов Т-лимфоцитам. Презентация сопровождается продукцией ИЛ-12, ФНО-α. После этого начинается процесс дифференцировки Т-лимфоцитов и выбора пути развития иммунного ответа в сторону либо CD4+ , либо CD8+ лимфоцитов.
Для того чтобы CD4+ Т-клетки активировались, необходимо наличие на их мембране определенных специфических мембранных рецепторов (CD28, CTLA-4/CD152 и CD-40L), которые будут комплементарно связываться с ко-стимулирующими молекулами (B7.1/CD80, B7.2/CD86, B7h/CD40) дендритных клеток. B7.1 и B7.2 могут взаимодействовать как с CTLA-4, так и с CD28.
Взаимодействие B7 с CD28 стимулирует выработку ИЛ-2, который индуцирует пролиферацию CD4+ Т-лимфоцитов. Если же B7 связывается с CTLA-4, то возникает препятствие для дальнейшего взаимодействия B7/CD28, что предупреждает продукцию ИЛ-2 и ингибирует процесс дифференцировки и пролиферации CD4+ Т-лимфоцитов.
У людей с генетической предрасположенностью имеется дефект гена CTLA-4. Нарушение функционирования этого рецептора ведет к нерегулируемому возбуждению CD4+ Т-лимфоцитов и развитию аутоиммунного процесса. Активированные В-клетки синтезируют аутоантитела класса G преимущественно к таким антигенным детерминантам, как тиреопероксидаза и тиреоглобулин [5, 19].
Низкая экспрессия рецептора CTLA-4 снижает возможность их активации Тreg. Что ведет к снижению контроля пролиферации CD8+ Т-лимфоцитов. Избыточное количество CD8+ Т-лимфоцитов ведет к избыточной продукции ИФ-γ. ИФ-γ, в свою очередь, стимулирует продукцию фолликулярными клетками ЩЖ хемокинов (CXCL9, CXCL10 и CXCL11), взаимодействующих исключительно с хемокиновым рецептором CXCR3, которые приводят к еще большей активации CD8+ Т-лимфоцитов [5, 10, 23].
Показано повышение риска развития АТ у людей с дефектом гена stat4, его продукт – фактор транскрипции STAT4, активирует транскрипцию генов в ответ на провоспалительные цитокины. Кроме того, было выяснено, что STAT4 участвует в развитии АТ у пациентов с псориазом [18].
На поверхности Т-лимфоцитов располагается гликопротеин CD3, экспрессия которого идет совместно с Т-клеточным рецептором (TКР). Роль CD3 заключается в обеспечении процесса адгезии Т-лимфоцитов с АПК. Гликопротеин CD3 состоит из нескольких цепей. Мутации в гене cd247 ведут к формированию нового варианта дзета-цепи CD3 рецептора, что влияет на сродство Т-лимфоцитов к аутоантигенам, стимулируя развитие аутоиммунного процесса, в частности в ЩЖ [5, 14].
Известны исследования, показывающие наличие взаимосвязи между микрофлорой кишечника синтезируемыми ею эндотоксинами, а также активностью эндотоксинового иммунитета с частотой развития ДТЗ [1].
Аутоиммунный процесс ведет к иммунному воспалению в тканях ЩЖ, ее дистрофии и дальнейшему фиброзу. Снижение синтеза тиреодных гормонов ведет к усилению выработки ТТГ по механизму обратной связи. ТТГ провоцирует рост эпителия щитовидной железы [14, 15, 23].
Большое влияние на продукцию ТТГ и активность тиреоидной гормональной оси оказывает гипоталамо-гипофизарно-надпочечниковая система, в связи с чем становится понятным возникновение дисфункций ЩЖ и нарушения продукции тиреоидных гормонов при стрессе [4, 21].
Кроме увеличения в крови больных CD95 рецепторных нейтрофилов, отмечается также повышение титра ФНО-α. ФНО-α является стимулятором апоптоза и потенцирует связывание Fas-рецептора с Fas-лигандом. ФНО-α в разных случаях способен не только стимулировать, но и блокировать развитие апоптоза [10, 13, 14].
Основное значение приобретает адапторный белок TRADD, который индуцирует апоптоз, но при его гиперэкспрессии, TRADD может активировать NF-kB фактор транскрипции, который блокирует запуск апоптоза. ФНО-α ведет к стимуляции экспрессии на нейтрофилах и клетках эндотелия факторов адгезии, рецепторов хемотаксиса, индукции синтеза цитокинов, фагоцитоза и цитотоксическому эффекту [10, 13, 14].
При АТ выявляются достаточно значимые изменения уровня цитокинов в крови больных. Увеличение титра ФНО-α прямо пропорционально активности воспалительного процесса. Повышение уровня ИФ-γ выявлялось чаще у больных с манифестирующим гипотиреозом, это может свидетельствовать о наличии тяжелых деструктивных процессов в ЩЖ и снижении ее компенсаторных возможностей. Отмечаются также высокие уровни ИЛ-4, ИЛ-8, что является достоверным признаком активного аутоиммунного процесса [3, 13].
Известно, что ИЛ-8 в несколько раз увеличивает экспрессию на поверхности нейтрофилов рецепторов адгезии семейства β2 интегринов и повышает их аффинность к связываемым лигандам (ICAM 1,2), усиливает адгезию и хемотаксис нейтрофилов в очаг воспаления. Связывание ИЛ-8 с его рецепторами, обильно представленными на нейтрофилах, приводит к активации сократительного аппарата клетки, образованию цитоплазматических ламелл и экзоцитозу гранулсодержащих лизосомальные ферменты. Кроме того, ИЛ-8 стимулирует бактерицидность нейтрофилов, потенцирует аттракцию фагоцитов к антигенным детерминантам и стимулирует синтез лейкотриенов [6, 10].
ИЛ-4 способен усиливать гуморальный иммунитет, за счет активации Тh-2 пути, с последующим образованием плазматических клеток и антител, преимущественно IgG. ИЛ-4 обладает как провоспалительным (симулирует высвобождение ФНО-α, ИЛ-1, ИЛ-8), так и противовоспалительное действие (за счет депрессии транскрипции генов и усиления деградации мРНК провоспалительных цитокинов) [6, 10].
При исследовании состояния иммунного статуса у больных c АТХ отмечалось уменьшение относительной концентрации CD8+ Т-лимфоцитов и снижение относительного и абсолютного содержания CD19+-клеток. Также у больных с АТХ снижено содержание в сыворотке IgG. Это говорит о том, что ведущую роль при АТХ играют антитела, которые в присутствии компонентов комплемента оказывают цитотоксическое действие и разрушают ткань ЩЖ [14, 15].
У больных ДТЗ установлено повышение процентного содержания лимфоцитов в периферической крови и абсолютного количества CD19+-клеток. А также повышение относительного и абсолютного уровней HLA-DR+-клеток. Состояние реактивности гуморального звена иммунной системы у больных ДТЗ характеризуется снижением уровня синтеза IgA, а также повышением концентрации циркулирующих иммунных комплексов в крови [14, 23].
При ДТЗ ЩЖ увеличена в размерах, отмечается относительное увеличение синтеза тиреоидных гормонов в начале заболевания. В исходе ДТЗ в гипертрофированных тиреоцитах начинают происходить дистрофические изменения, что в своем исходе ведет к фиброзу ЩЖ, как и при АТХ [14, 23].
Несмотря на схожий патогенез этих заболеваний, имеются явные различия в иммунных изменениях при ДТЗ и АТХ. Аутоиммунный процесс при ДТЗ характеризуется повышением количества цитотоксических клеток, В-лимфоцитов. Высокое количество В-клеток ведет к повышенной продукции IgG при ДТЗ. При АТХ таких изменений не наблюдается.
Следовательно, несмотря на общую роль В-лимфоцитов в течении АТХ и ДТЗ, отличительной чертой патогенеза ДТЗ является участие CD8+-клеток, IgМ и IgG. Возможно, данные особенности принимают участие не только в формировании различного патогенеза, но и разных морфологических и функциональных характеристик ЩЖ [23].
Особое внимание хотелось бы еще раз уделить тем иммунологическим особенностям, которые возникают на фоне АТ и их патологическому влиянию на организм пациента. Было выявлено, что при патологии ЩЖ активируется иммунная система, что проявляется высокими уровнями ФНО-α, ИЛ-8, ИЛ-1 и ИЛ-4.
Интерлейкин-4 – основными продуцентами этого цитокина являются лимфоциты, тучные клетки, эозинофилы и базофилы. Основной функцией ИЛ-4 является стимулирование развития иммунного ответа по Тh2 пути. Кроме того, ИЛ-4 снижает выделение провоспалительных цитокинов (ФНО-α, ИЛ-1, ИЛ-8) и простагландинов из активированных моноцитов, способен ингибировать синтез цитокинов ТН1 лимфоцитами (ИЛ-2, ИФ-γ и др.). ИЛ-4 играет важную роль в развитии реакций гиперчувствительности I типа, так как индуцирует переключение синтеза IgG1 на IgG4 и IgE и индуцирует выброс биологически активных молекул тучными клетками [6, 10].
Высокие значения ИЛ-1 также неслучайны. Клетки-продуценты моноциты, макрофаги, АПК, NK-клетки, В-лимфоциты и другие. ИЛ-1 вызывают воспалительные сосудистые реакции, повышение температуры тела и индукцию секреции белков острой фазы. Мишенями для действия этого цитокина являются эндотелиоциты (сосудистая воспалительная реакция), гипоталамус (ИЛ-1 действует на центр терморегуляции, этим объясняется повышение температуры тела у больных при гипертиреозе), а также печень (выработка белков острой фазы и поддержание иммунного воспаления) [6, 10].
ФНО-α продуцируется макрофагами, моноцитами, NК-клетками и Т1-клетками. Это провоспалительный цитокин, стимулирует сосудистые реакции, индуцирует выработку белков острой фазы, активирует нейтрофилы [6, 10].
АТ имеют множество негативных последствий для всего организма. Однако мы привыкли рассматривать его последствия только со стороны неблагоприятного влияния гипо- либо гипертиреоза. Выше описывались иммунные нарушения, которые возникали на фоне АТ, поэтому рассматривая осложнения АТ, важно помнить, что они вызваны не только гормональными, но и иммунными нарушениями.
Таким образом, аутоиммунные тиреопатии – это заболевания с наследственной предрасположенностью, в основе которых лежит иммунное воспаление щитовидной железы, проявляющееся изменениями тиреоидного статуса, как в сторону гипер-, так и гипотиреоза, при этом клинические проявления данных заболеваний не исчерпываются нарушениями тиреоидного статуса, большой вклад в патогенез данных нозологий вносят вторичные нейроиммунноэндокринные нарушения как отражение существующего аутоиммунного процесса.
Читайте также:
- Когда прекращается понос у детей при кишечной инфекции
- Что нужно взять с собой в инфекционную больницу с ребенком
- Инфекционная больница туапсе отзывы
- Какие скрытые инфекции проявляются только у женщин
- Инфекционная больница кропоткина лаборатория
 Пожалуйста, не занимайтесь самолечением!При симпотмах заболевания - обратитесь к врачу.
Пожалуйста, не занимайтесь самолечением!При симпотмах заболевания - обратитесь к врачу.
Copyright © Иммунитет и инфекции