
Где лечат губчатый энцефалит
Воспалительный процесс, охватывающий ткань головного мозга, а также часто мозговую оболочку и субарахноидальное пространство, вызванный наличием микроорганизмов в ткани головного мозга (также как дальнейшее развитие менингита).
1. Этиологический фактор: обычно вирусы, чаще всего: вирус клещевого энцефалита (КЭ) →разд. 18.5.2, ВПГ или ВВЗ (в других регионах флавивирусы); редко: вирус кори, паротита, краснухи, СМВ, энтеровирусы (тип 71), бешенства, ВИЧ, ЭБВ, вирус гриппа, ВГЧ-6, грибы ( Candida , Cryptococcus neoformans , Aspergillus ), простейшие — амебы ( Naegleria fowleri , Acantamoeba spp. , Balamuthia mandrillaris ).
2. Источник инфекции и путь передачи (в зависимости от этиологического фактора; резервуар — обычно люди, только в случае бешенства дикие животных (лисы, белки, летучие мыши) и собаки, реже кошки; в случае амеб — загрязненная вода. Путь инфицирования — в зависимости от возбудителя воздушно-капельным путем или при непосредственном контакте с больным (или выделениями больного), переносчики, в случае бешенства укус больного животного или непосредственный контакт поврежденной кожи или слизистых оболочек со слюной животного.
3. Эпидемиология: заболеваемость вирусным энцефалитом ≈1,6/100 000/год (550–650 случаев в год). Сезонность зависит от типа вируса (КЭ, ВВЗ, энтеровирусы). Факторы риска: пребывание в закрытых сообществах, пользование общественными банями и бассейнами, агаммаглобулинемия, нарушение клеточного иммунитета, контакт с бездомными и дикими животными (бешенство), пребывание в эндемичных по КЭ районах, контакт с больным; факторы риска грибковых инфекций ЦНС →разд. 18.6.1.
КЛИНИЧЕСКАЯ КАРТИНА И ЕСТЕСТВЕННОЕ ТЕЧЕНИЕ наверх
Во многих случаях вначале продромальные симптомы (гриппоподобные, диарея, лихорадка, лимфаденопатия) и/или симптомы основного заболевания (напр. кори, эпидемического паротита, ветряной оспы). Особенно тяжелое и динамическое течение имеет герпетический энцефалит (обычно нет высыпаний простого герпеса на коже или слизистых оболочках). В клинической картине доминируют спутанность сознания , лихорадка различной степени тяжести и очаговые симптомы: расстройства сознания качественные (психотические синдромы, расстройства личности) и количественные (снижение сознания до глубокой комы включительно); головная боль, тошнота и рвота, брадикардия (симптомы отека мозга и повышенного внутричерепного давления →разд. 2.29); припадки судорог очаговых и общих; парезы и спастические параличи, также другие симптомы вовлечения пирамидного тракта (синдром повреждения центрального двигательного нейрона); парезы и параличи черепно-мозговых нервов (чаще III, VI, IV и VII); вялые параличи (свидетельствуют о поражении ствола головного мозга); мозжечковые симптомы (чаще всего в течении ветряной оспы — воспаление мозжечка); ухудшение памяти, даже глубокие амнестические синдромы; афазия, наиболее часто двигательного типа или смешанного; вегетативные симптомы — чрезмерная потливость, чередование замедления и ускорения частоты сердечного ритма, гипотермия и гипертермия, слюнотечение и гидрофобия (напр. при бешенстве).
При бешенстве может развиться энцефалит, который клинически может проявляться психо-двигательным возбуждением (возбужденная форма) или вялым параличем (паралитическая форма). При возбужденной форме наблюдаются приступы психомоторного возбуждения, галлюцинации, нарушения сознания; в период между приступами пациент находится в сознании. Приступы могут провоцироваться внешними факторами — акустическими, зрительными или тактильными раздражителями. Они могут быть осложнены появлением общих судорог, остановкой дыхания и острой остановкой кровообращения. В этой фазе бешенства наблюдаются также патогномоничные симптомы: водобоязнь и аэрофобия, при которых доходит до резких сокращений диафрагмы и вспомогательных дыхательных мышц. У некоторых пациентов фазы возбуждения нет, а в клинической картине доминирует вялый паралич.
Дополнительные методы обследования
1. MРТ (предпочтительно) или КТ головы: обязательно у каждого больного с подозрением на энцефалит. Изменения можно увидеть уже на ранних стадиях (особенно на MРТ), их локализация и характер могут указать на возможную этиологию инфекции или помочь исключить другие причины неврологических симптомов. В острой фазе — признаки отека головного мозга.
2. Общий анализ СМЖ →разд. 27.2: только повышенное давление, незначительное увеличение количества мононуклеарных клеток и/или концентрации белка. Когда сопровождается менингитом, в СМЖ изменения зависят от этиологии. Повышенное внутричерепное давление (напр. отек мозга) является противопоказанием для выполнения люмбальной пункции →разд. 24.12; если имеются симптомы отека мозга или очаговые → показания для люмбальной пункции на основании МРТ или КТ.
3. Микробиологические исследования: как при менингите →разд. 18.7.1; диагностика КЭ →разд. 18.6.2; в случае подозрения неглериоза — микроскопическое исследование неотцентрифугированной жидкости (окрашивание методом Гимза, PAS или гематоксилином и эозином; визуализация подвижных амеб). Основой для определения этиологии вирусных инфекций ЦНС является обнаружение вирусного генетического материала в спинномозговой жидкости с помощью ПЦР или ОТ-ПЦР; если клиническая картина указывает на герпетический энцефалит, а результат ПЦР отрицательный → необходимо рассмотреть повторное исследование через 3–7 дней. В случае флавивирусной инфекции продолжительность виремии недолгая (2–7 дней после заражения), поэтому молекулярные исследования в этом случае имеют ограниченное применение (в основном используются серологические тесты). При диагностике бешенства: определение антигенов, молекулярные исследования, биопробы; вирус может быть выделен из слюны, цереброспинальной жидкости, мочи, посмертно из ткани головного мозга.
4. Серологические исследования (не применяют у больных с ослабленным иммунитетом): специфические IgM в СМЖ (диагностика КЭ, ВПГ, ВВЗ, ЭБВ), при необходимости, специфические IgG в СМЖ и сыворотке (концентрация в СМЖ, которая в 20 раз превышает концентрацию в сыворотке, является подтверждением инфекции ЦНС). Результат обычно отрицательный на протяжении первых 1–2 нед. болезни.
5. ЭЭГ: показана всем пациентам. Довольно специфичная картина ЭЭГ при ВПГ-энцефалите (часто опережает те изменения, которые получают при нейровизуализации).
Менингит, опухоль головного мозга (абсцесс, субдуральная эмпиема, внутричерепная гематома, новообразование первичное или метастатическое, цистицеркоз и эхинококкоз головного мозга), инсульт или субарахноидальное кровоизлияние, церебральный васкулит (изолированный или в процессе системных заболеваний), нарушения обмена веществ (гипогликемия или гипергликемия, гипонатриемия, гипокальциемия), интоксикации (лекарства, наркотики), энцефалопатия печеночная или уремическая, эпилепсия и эпилептический статус, психозы, т. н. параинфекционный (постинфекционный) энцефалит (аутоиммунный процесс, сопровождающий вирусное заболевание [напр. корь или ветряную оспу], или, очень редко, связанный с некоторыми прививками [напр. против бешенства или кори], вызывает мультифокальную демиелинизацию, характеризующийся обычно легким или средней тяжести течением [низкая смертность]; в СМЖ не обнаруживают наличие вирусов; необходимо отличить от рассеянного склероза; основное диагностическое значение имеет MРТ).
1. Ацикловир в/в 10 мг/кг каждые 8 ч (правила инфузии →гл. 18.1.6) — необходимо применить эмпирически как можно скорее во всех случаях энцефалита, особенно при тяжелом течении, не дожидаясь вирусологического подтверждения (эффективность при герпетическом энцефалите тем выше, чем раньше начато лечение). Продолжать лечение в течение 3 нед.
2. В обоснованных случаях подозрения или подтверждения конкретной этиологии энцефалита необходимо рассмотреть следующее:
1) ЦМВ → ганцикловир в/в (5 мг/кг инфузии каждые 12 ч в течение 3 нед.) возможно в сочетании с фоскарнетом в/в (60 мг/кг массы тела каждые 8 ч или 90 мг/кг каждые 12 ч); также необходимо рассмотреть применение ганцикловира у больного с нарушением клеточного иммунитета с энцефалитом неизвестной этиологии;
2) вирус Варицелла-Зостер (ВВЗ) → ацикловир в/в 10–15 мг/кг каждые 8 часов на протяжении 10–14 дней (альтернативно ганцикловир);
3) вирус герпеса человека 6-го типа (ВГЧ-6) у пациента с нарушениями клеточного иммунитета → ганцикловир или фоскарнет.
4) грибковые инфекции →1146.
5) неглериоз амфотерицин В 1,5 мг/кг/сутки в/в (±1,5 мг/сутки интратекально) + рифампицин 10 мг/кг/сутки + флуконазол 10 мг/кг/сутки в/в или п/о + милтефозин 50 мг каждые 8 часов п/о.
Как при менингите →разд. 18.6.1. Имеет важное значение.
Разрешение симптомов и улучшение общего состояния больного свидетельствуют об эффективности лечения. Регулярное контрольное исследование СМЖ не обязательно. Если нет улучшения или появление осложнений → необходимо повторное исследование СМЖ и визуализирующие исследования мозга (оптимально МРТ). Поствоспалительные изменения в ликворе могут сохраняться еще длительное время после разрешения острой фазы заболевания.
В острой фазе — эпилептический статус, вклинение мозга (последствие повышенного внутричерепного давления), СНАДГ →разд. 8.2. Поздние осложнения — постоянные парезы и параличи, эпилепсия, психотические расстройства, нарушения памяти, состояния слабоумия, афазия.
При энцефалите прогноз неблагоприятный, особенно высокая смертность при герпетической инфекции (без специфического лечения 70–80 %, если лечение было начато во время, до потери сознания — 30 %). Воспаление головного мозга или мозжечка во время ветряной оспы →1092; микозы ЦНС →1147. Смертность при заражении N. fowleri , Acantamoeba sp. и B. mandrillaris составляет >95 %.
1. Вакцинация: против кори, эпидемического паротита и краснухи, ветряной оспы, гриппа, полиомиелита, клещевого энцефалита, бешенства и вируса японского энцефалита.
2. Пассивная иммунопрофилактика : в конкретных ситуациях специфический иммуноглобулин против ветряной оспы (VZIG), бешенства (RIG) или гаммаглобулин для профилактики кори.
1. Избегать контакта с дикими животными и бродячими собаками и кошками (профилактика бешенства).
2. Неспецифические методы защиты от клещей →разд. 18.5.1 (профилактика КЭ).
3. Обязанность сообщения в органы исполнительной власти региона в сфере здравоохранения и управления Роспотребнадзора по субъекту Федерации : да.
(Подострая губчатая энцефалопатия)
, MD, Case Western Reserve University
Last full review/revision December 2018 by Pierluigi Gambetti, MD
БКЯ имеет три формы (1):
сБКЯ является наиболее распространенным типом, на который приходится около 85% случаев. сБКЯ обычно проявляется у пациентов в возрасте >40 лет (медиана – около 60 лет).
Семейная БКЯ диагностируется в около 5 до 15% случаев. Наследование аутосомно-доминантное, возраст начала заболевания, как правило, раньше, чем при сБКЯ с большей длительностью заболевания.
Приобретенная БКЯ вероятно, объясняет
Вариантная БКЯ (вБКЯ)
ВБКЯ редкая приобретенная форма БКЯ. Большинство случаев произошло в Великобритании (UK), которые составили 178 случаев в сентябре 2018 года, по сравнению с 53 случаями во всех других европейских и неевропейских странах. вБКЯ диагностировалась после приема в пищу мяса, полученного от крупного рогатого скота, заболевшего губкообразной энцефалопатией крупного рогатого скота (ГЭКРС), также называемой коровьим бешенством.
В случае заражения вБКЯ, симптомы развиваются в более раннем среднем возрасте ( 30 лет), чем в случае заражения сВКЯ. В недавно диагностированных случаях инкубационный период (время между употреблением в пищу зараженной говядины и развития симптомов) составлял от 12 до более чем 20 лет.
В начале 1980-х годов из-за слабо контролируемых правил переработки побочных продуктов животного происхождения, зараженной ткани, вероятно, от овец, зараженных скрепи, или крупного рогатого скота, зараженных ГЭКРС, скрепи прионового белка (PrP Sc ) попадали в корм для крупного рогатого скота. У сотен тысяч голов крупного рогатого скота развилась ГЭКРС. Несмотря на широкое воздействия, относительно у немногих людей, которые употребляли в пищу мясо больного крупного рогатого скота развилась вБКЯ.
В связи с длительным инкубационным периодом ГЭКРС связь между заболеванием и зараженным мясом в Великобритании не была установлена до тех пор, пока заболеваемость ГЭКРС не переросла в эпидемию. Эпидемия ГЭКРС перешла под контроль после массивного убоя скота и после изменения в процедурах производства технических фабрикатов, которые резко сократили загрязнения мяса тканями нервной системы. В Великобритании ежегодное число новых случаев вБКЯ, которое достигло пика в 2000 году, неуклонно сокращалось, и было зафиксировано только 2 случая после 2011 года.
Четыре случая вБКЯ были связаны с переливанием крови, они диагностировались у людей, которым была проведена трансфузия между 1996 и 1999. В Великобритании приблизительно 1 из 2000 человек могут быть носителями вБКЯ (на основании изучения большого количества образцов ткани аппендикса), но не имеют никаких симптомов; эти люди могут передавать болезнь, являясь донорами крови или при проведении хирургической процедуры. Таким образом, неясно, имеется ли группа пациентов, которым переливали инфицированную кровь и которые, таким образом, находятся в зоне риска последующего развития вБКЯ. Тем не менее, новые направления в критериях, связанных с донорством, связанные с вБКЯ, могут дополнительно снизить риск передачи вБКЯ при переливании крови, который уже достаточно низкий за пределами Франции и Великобритании.
Хотя нет официальных данных ни об одном случае возникновения вБКЯ в Северной Америке, были опубликованы сообщения о случаях ГЭКРС у нескольких особей крупного рогатого скота в Северной Америке (4 в США и 19 в Канаде).
Общие справочные материалы
2. Ritchie DL, Barria MA, Peden, AH, et al: UK Iatrogenic Creutzfeldt-Jakob disease: Investigating human prion transmission across genotypic barriers using human tissue-based and molecular approaches. Acta Neuropathol 133 (4): 579–595, 2017. doi: 10.1007/s00401-016-1638-x.
3. Cali I, Cohen ML, Haik S, et al: Iatrogenic Creutzfeldt-Jakob disease with amyloid-β pathology: An international study. Acta Neuropathol Commun 6 (1):5, 2018. doi: 10.1186/s40478-017-0503-z.
Клинические проявления
Приблизительно у 70% пациентов, заболевших БКЯ, выявляются нарушение памяти, внимания и изменение поведения, которые в конечном счете развиваются у всех пациентов; у 15–20% отмечается расстройство координации и атаксия, которые часто появляются на ранних стадиях заболевания. На более поздних стадиях могут возникнуть миоклонические судороги, вызываемые громким звуком (миоклония при испуге) или другими сенсорными стимулами. У людей с вБКЯ диагностируются психические симптомы (например, тревога, депрессия), а не потеря памяти. Более поздние симптомы похожи при обеих формах.
Помимо наиболее характерных для БКЯ деменции, атаксии и миоклонических судорог могут появиться и другие неврологические расстройства (например, галлюцинации, эпилептиформные припадки, нейропатия, различные двигательные нарушения).
При сБКЯ часто встречаются зрительные нарушения (например, дефекты поля зрения, диплопия, затуманенность или нечеткость зрения, зрительная агнозия).
Губчатая энцефалопатия, также известная под названием синдром Кройцфельда-Якобcа и vCJD, до определённого момента встречалась крайне редко.
А точнее, что это одна и та же болезнь, опасная и для человека, и для коров в равной степени.
И в это же время в Альбионе случилась настоящая эпидемия коровьего бешенства, и виной тому стала сельскохозяйственная программа, принятая ещё правительством Маргарет Тэтчер.
Кому-то из чиновников от сельского хозяйства пришла в голову мысль сделать травоядных коров. каннибалами. В корм крупному рогатому скоту стали добавлять муку, полученную из перемолотых костей мёртвых коров.
И всё, казалось бы, замечательно: корм становится более питательным и калорийным, да и останки забитой скотины идут в дело.
Однако это привело к катастрофе, истинные масштабы которой человечеству, возможно, ещё только предстоит постигнуть.

Таким образом, если мутировавший прион попадает в гамбургер из ближайшего Fast-Foodа (и таковых в Британии чуть ли ни по три штуки на каждом углу), то употребивший этот гамбургер человек также имеет все шансы получить vCJD.
Инкубационный период болезни — около 10-15 лет. Болезнь по-прежнему считается неизлечимой. Однако время идёт, и множество учёных ищет способы с ней управиться.
Когда в клетке начинается синтез неправильных прионов, внутриклеточная система контроля должна их уничтожать, но при vCJD она, очевидно, оказывается нарушенной. И прионы-убийцы накапливаются внутри клетки.
Поражённая клетка умирает ещё до того, как неправильные прионы внутри них начинают образовывать комки.
Как выяснилось, непосредственным убийцей клетки являются как раз оказавшиеся в меньшинстве нормальные белки. Очевидно, чтобы не допустить дальнейшего синтеза и распространения прионов-убийц. Если неправильные прионы начинают превалировать в организме, гибель нервных клеток начинает происходить в массовом порядке.
Лечение, а точнее, профилактика
Сотрудники Имперского колледжа в Лондоне (Imperial College in London) обнаружили, что у мышей это заболевание можно остановить, а точнее — предотвратить, с помощью так называемых моноклональных антител — молекул, вырабатываемых иммунной системой живого организма.
Эти молекулы связываются и с нормальными прионами, и с неправильными, и мешают им связываться между собой. А соответственно, препятствуют превращению нормальных прионов в неправильные.
Те мыши, которым вводили моноклональные антитела, не выказывали никаких признаков заболевания даже через год, после того, как мыши, которым не давали вакцину, умерли.
Однако, те мыши, которым давали моноклональные антитела, ещё не выказывали никаких признаков заболевания. Они были, условно говоря, заражены, но ещё не заболели.
Помимо двух упомянутых групп мышей существовала ещё и третья. Этим грызунам антитела не вводили до тех пор, пока они не начали выказывать признаков заболевания в полной мере.
Как и следовало ожидать, на них лечение не оказало никакого воздействия.
Сотрудники Имперского колледжа, однако, собираются продолжать эксперименты в этом же направлении. На следующем этапе планируется увеличить концентрацию моноклональных антител, вводимых в мозг, чтобы они замедляли развитие болезни даже тогда, когда она уже начинает одолевать организм.

Кроме того, планируется произвести генетическую модификацию моноклональных антител, чтобы они больше напоминали антитела, встречающиеся в человеческом организме. Это позволит использовать их при клинических испытаниях.
Возглавляющий эти исследования специалист, доктор Саймон Хоук (Simon Hawke), предупредил, что понадобится ещё немало времени и сил, прежде чем появится что-то похожее на лекарство для человека.
Более того, это будет не восстанавливающее, а лишь профилактическое средство.
К тому же понадобятся надёжные средства диагностики vCJD, а их-то как раз пока и нет.
В общем и целом, команда Имперского колледжа не первая, кто достигает каких-либо успехов на поприще борьбы с vCJD. Более того, испытания на людях уже имеют место быть.
Напомним, что в конце 2002 года Верховный суд Великобритании дал разрешение на введение экспериментального препарата против губчатого энцефалита прямо в мозг двум подросткам, умирающим от этого страшного заболевания.
Двое подростков — 18-летний юноша и 15-летняя девушка — сейчас пребывают уже в слишком тяжёлом состоянии, чтобы понимать, что происходит вокруг. Их родители дали добро на испытание препарата, который должен блокировать дальнейшее производство приона-убийцы в организме.
Этот препарат — полисульфат пентозана — используется в медицине уже сорок лет. Это лекарство принимают страдающие от цистита и абдоминальных болей.
Более ранние исследования, проведённые в Японии и Великобритании, продемонстрировали способность этого препарата останавливать развитие скрэпи в организмах мышей.
Как видим, стартовая полоса на пути к лечению болезни давно пройдена. Учёные постепенно осваивают методы профилактики заболевания, а при большой удаче — смогут её останавливать, когда болезнь начинаются уже поражения мозга.
Проблема в том, что после определённого момента бороться с этим заболеванием уже бесполезно: восстановление повреждённых нервных тканей — причём, не важно, как и чем именно повреждённых — один из проклятых вопросов медицины, на который пока внятного ответа не существует.
Как не существует и надёжной заблаговременной диагностики vCJD.
Остаётся добавить, что из Великобритании уже приходят обнадёживающие известия: статистика указывает на уменьшение количества случаев vCJD в последние два года. Но действительно ли это признак того, что эпидемия спадает, так, по сути, и не начавшись, — никто пока сказать не может.
Инфекционные болезни (общее учение)
Прионовые инфекции
Лекция курса патологической анатомии профессора В.Г. Шлопова
Болезнь Крейтцфельда-Якоба (БКЯ)
Болезнь Крейтцфельда-Якоба представляет собой подострую энцефалопатию, которая характеризуется медленной прогрессирующей гибелью нейронов. Болезнь проявляется обычно у взрослых и характеризуется быстрым развитием деменции, которая сопровождается пирамидальными и экстрапирамидальнами симптомами. Заболевание является спорадическим и встречается с частотой 1:1000000; реже встречаются семейные и ятрогенные случаи заболевания. В 1968 году была доказана трансмиссивная (инфекционная) природа этого заболевания у приматов.
Заражение происходит при употреблении в пищу мяса коров, больных аналогичным заболеванием. Случаи передачи от человека к человеку были описаны при имплантации внутричерепных электродов, пересадке роговицы и, наиболее часто, при введении гормонов роста, экстрагированных из гипофиза человека.
В мозге пораженных наиболее часто наблюдается диффузная атрофия коры головного мозга с губкоподобными изменениями, особенно в неокортексе и распространенной дистрофией нейронов. На разрезе видны очаги размягчения вещества мозга, иногда в виде полостей, выполненных мутноватым серовато-розовым кашицеобразным содержимым. Микроскопически обнаруживается уменьшение количества нейронов и реактивная пролиферация астроцитов. В отростках нейронов и астроцитов обнаруживаются многочисленные маленькие вакуоли, из-за чего появился термин "губчатый энцефалит". При данном заболевании в ткани мозга не обнаруживаются признаки воспалительного ответа.
Болезнь Крейтцфельда-Якоба представлена тремя классическими формами:
- спорадическая форма (85-90% всех случаев);
- семейная форма (10-15%);
- ятрогенная форма (% еще окончательно не установлен).
Кроме того, по предложению британских исследователей, в настоящее время выделена еще одна форма, так называемая болезни Крейтцфельда-Якоба (nv-CJD).
Спорадическая форма болезни Крейтцфельда-Якоба относится к числу редких заболеваний. Она распространена на всех континентах. Средний уровень регистрируемой в мире заболеваемости БКЯ составляет около 0,5-1,0 на 1 млн жителей. БКЯ встречается в разных возрастных группах - от 17 до 82 лет, в зависимости от формы болезни. Диапазон продолжительности течения болезни колеблется от нескольких недель до восьми лет. Однако, средняя продолжительность жизни от начала болезни - шесть месяцев.
Ятрогенные формы являются следствием нейрохирургического заражения через недостаточно обеззараженный хирургический инструментарий или электроды, при трансплантации роговицы, твердой мозговой оболочки или при лечении дериватами гипофиза человека ( гормоны роста и гонадотропины ). Инкубационный период ятрогенной, как, вероятно, и других форм БКЯ, зависит от очень многих факторов: от способа и ворот поступления инфекта в организм, его фенотипа, дозы инфекта, от генотипа реципиента и т.д. В тех случаях, когда внедрение агента происходило непосредственно в центральную нервную систему, инкубационный период составлял от 10 до 30 месяцев и первыми признаками в клинике была деменция . В то время, когда инфекция поступала в организм из периферии, например, при введении гормонов роста или гонадотропинов, инкубационный период удлинялся до 5 лет и более, достигая иногда 35 лет. Эти больные страдали исключительно мозжечковой атаксией.
6 апреля 1996 года в журнале "The Lancet" были опубликованы две научные статьи, которые сразу привлекли внимание не только ученых, но и широкой общественности. В одной из них приведены два случая развития заболевания Крейтцфельда-Якоба у двух молодых людей с необычной клинической картиной. Изучение генотипа этих больных показало, что оба они гомозиготы с кодоном 129 гена прионового белка PrP таким же, как в большинстве спорадических или ятрогенных форм болезни Крейтцфельда-Якоба. Эта группа больных отличалась по своим клиническим симптомам от больных спорадической формой БКЯ. После эксперимента на мышах на основании анализа длительности инкубационного периода и характера наблюдаемых анатомических повреждений, высказано предположение, что все эти случаи, по-видимому, от одного родоначальника прионов, по своей природе соответствуют приону овечьей лихорадки. Авторы статей пришли к заключению, что наблюдаемый ими необычный вариант болезни Крейтцфельда-Якоба, поражающий более или менее молодой возраст, связан с новым фактором риска. И таким фактором, возможно, является прионовая болезнь коров (СЭК). В 1996 было предложено выделить новую форму этой болезни и обозначить ее как болезни Крейтцфельда-Якоба.
Клинические симптомы болезни Крейтцфельда-Якоба (nv-CJD) ближе к болезни Куру и ятрогенным формам БКЯ. У этих больных отмечено выраженное преобладание атаксии над деменцией и большое количество прионовых амилоидных бляшек с PrP в биопсийном материале головного мозга. Бляшки моноцентрические, такие же, как при болезни Куру, но они окружены характерной спонгиозной зоной. Время развития болезни от момента первых клинических проявлений до их завершения составляет около 12 месяцев. Специфического лечения не существует и болезнь имеет 100% летальность.
С появлением Интернета и свободных СМИ, люди стали узнавать всё больше о смертельных заболеваниях — инфекционных, вирусных, онкологических и наследственных. Но мало кто слышал о фатальных прионных болезнях. Несмотря на клинические испытания, на данный момент не существует ни одного доказанного универсального лечения этой группы заболеваний. Невролог Ричард Джонсон из Университета Джона Хопкинса говорит, что если прионы пациента превратились в патологические, он умирает, и мы не можем этого избежать.
Прионные заболевания — они же называются трансмиссивными губчатыми энцефалопатиями — представляют собой семейство редких прогрессирующих нейродегенеративных заболеваний, которые поражают как людей, так и животных. Их отличают:
- длительный инкубационный период;
- характерные губчатые разрыхления мозговой ткани, связанные с потерей нейронов;
- неспособность иммунной системы отреагировать на заражение, инициируя воспалительный процесс.
Прионные болезни поражают как людей, так и животных, быстро прогрессируют и всегда приводят к летальному исходу.
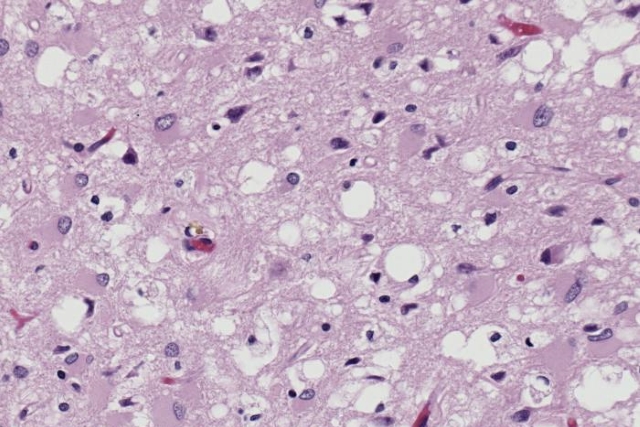
Возбудитель болезни — прионы — тип белков с аномальной третичной структурой, не содержащий нуклеиновых кислот. Сам термин относится к патологическим патогенным агентам, которые способны вызывать аномальное сворачивание специфических нормальных клеточных белков, которые называются как раз прионными белками, встречающихся чаще всего в мозге. Функции этих нормальных прионных белков до сих пор полностью не изучены.
Болезнь Крейтцфельдта – Якоба — БКЯ, псевдосклероз спастический, синдром кортико-стриоспинальной дегенерации, трансмиссивная спонгиоформная энцефалопатия, коровье бешенство.
Это прогрессирующее дистрофическое заболевание коры большого мозга, базальных ганглиев и спинного мозга. Считается основным проявлением губчатой энцефалопатии (прионной болезни). Излечение невозможно. БКЯ поражает людей всех национальностей и рас, мужчин и женщин, взрослых и детей.
Прионные белки — нормальные белки, которые присутствуют у каждого человека. Но есть определённые группы людей, у которых присутствует генетическая мутация, предрасполагающая их к синтезу патогенного прионного белка. Прионные болезни также могут передаваться путём прямого заражения, передача может произойти и в ходе хирургических манипуляций, использования человеческого гормона роста или употреблении заражённого мяса. Такой вид заражения называется ятрогенным и он остаётся в процентном меньшинстве относительно иных форм БКЯ.
Процентное соотношение ятрогенных случаев болезни Крейтцфельдта – Якоба в исследовании National CJD Research & Surveillance Unit у 177 пациентов.
- Гормон роста (соматотропин) — 53,1% (94 случая).
- Твердая мозговая оболочка (в том числе поедание) — 38,9% (69 случаев).
- Гонадотропный гормон — 2,25% (4 случая).
- Нейрохирургический инструментарий — 2,25% (4 случая).
- Пересадка роговицы — 1,69% (3 случая).
- Электроды для стереоэлектроэнцефалографии — 1,12% (2 случая).
- Пересадка печени — 0,56% (1 случай).
Есть случаи заражения, которые не классифицируются ни по одной из двух вышеупомянутых причин, в таком случае они считаются спорадическими, то есть возникшими спонтанно и самостийно, по независящим от генетики или внешних факторов обстоятельствам.
Доктор Ойбек Тургунхужаев, руководитель направления нейрореабилитации Междисциплинарного центра реабилитации (Москва), говорит, что окончательный диагноз человеку с подозрением на какое-либо прионное заболевание основывается на оценке клинических признаков и симптомов и ряде вспомогательных исследований. Долгое время единственным методом подтверждения диагноза была электроэнцефалография. Но поскольку общая чувствительность этого метода ограничена, полезность этого исследования была поставлена под сомнение.
Прионные заболевания неизлечимы, они неизбежно фатальны. Кроме этого, проблема заключается в том, что для постановки достоверного диагноза необходимо проводить вскрытие. Любое вскрытие — это риск для патологоанатома, так как были случаи ятрогенного заражения специалистов от умерших пациентов.
По приказу Роспотребнадзора, о том, что человек заболел прионной болезнью необходимо извещать в течение двух часов. При этом установление такого диагноза ведет за собой, по российским инструкциям, утилизацию всего оборудования, с которым пациент был в контакте. Именно поэтому, когда Медуза рассказывала случай одной из больных БКЯ, все клиники говорили о том, что у них нет оборудования для наблюдениях таких пациентов. На самом деле — это просто способ не потерять миллионы рублей, утилизируя даже аппарат МРТ. Если бы речь шла о сотнях поставленных диагнозов прионной болезни (например, в США регистрируется 300 случаев ежегодно, возможно, их больше), тогда речь шла бы о потери миллиардов рублей для российских больниц и бюджета. Именно поэтому официально диагноз не ставится, врачи не хотят об этом говорить, так как никакого официального распоряжения не существует, что диагноз ставить нельзя. В итоге выходит, что заболевание есть, смерти есть, а причины для родственников и умирающих людей — нет.
Им никто не скажет, что скорее всего родственники уже заразились. Никто не скажет, что нельзя пробовать сырой фарш или есть сырое мясо, тем более мозги. Также как из-за того, что диагноз не ставится, можно случайно пересадить орган больного прионной болезнью, тем самым заразив другого человека. Также это может произойти через хирургический инструмент (такие случае были, об этом ниже).
Когда мы просили хоть кого-то рассказать нам о прионных болезнях, практически никто не готов был говорить открыто. Так мы анонимно поговорили с врачом-неврологом одной из крупнейших московских больниц. «С прионами две проблемы. Во-первых, для постановки достоверного диагноза необходимо проводить вскрытие. Хотя формально (по российским руководствам, например) проводить вскрытие можно, хотя и в особенных условиях. Любое вскрытие — это, естественно, дополнительный риск для патологоанатомов, потому что были описаны случаи заражения патологоанатомов от умерших пациентов. никто не хочет переводить на них риск.
Во-вторых, так как прионные инфекции — это тяжело протекающие, неизлечимые заболевания (хотя и с довольно сложным путем передачи), в нашей стране чертовски сложные законы для регистрации и ведения таких пациентов; о случаях обязаны сообщать в случае выявления чуть ли не в течение двух часов, после постановки диагноза необходимо проводить уничтожение части в том числе дорогостоящего оборудования, которое, как может оказаться по факту, даже рядом не лежало с пациентом, нужно переоформлять документы и так далее.
Болезнь Крейтцфельда – Якоба (БКЯ) является одной из разновидностей прионных болезней. Это быстро прогрессирующее, фатальное нейродегенеративное заболевание, которое, как полагают, вызвано аномальной изоформой прионного белка. БКЯ встречается во всем мире, и согласно статистике, во всем мире заболевает 1 из миллиона человек.
Прионные болезни не идут по одному и тому же сценарию, у людей, страдающих одним и тем же прионным поражением могут разниться эпидемиология и патогенез. Болезнь Крейтцфельда-Якоба делят на несколько типов.
Спорадическая Болезнь Крейтцфельда-Якоба (сБКЯ) — наиболее распространенный вид трансмиссивных губчатых энцефалопатий человека, на долю которого приходится около 85% случаев зарегистрированных заболеваний прионной природы. СБКЯ имеет очень быстрое течение болезни — средняя продолжительность жизни после проявления признаков составляет всего шесть месяцев. Более 90% пациентов умирают в течение года после появления симптомов. Пик заболеваемости приходится на пожилых людей возрастом 60–70 лет, в других возрастных группах случается куда реже. Одной из гипотез происхождения сБКЯ является мнение, что это спонтанное нейродегенеративное заболевание, возникающее в результате соматической мутации гена PRNP или случайного структурного изменения в белке PrP, вызывающее образование PrPSc2. Эпидемиологические исследования не выявили связи спорадической формы БКЯ с экологическими факторами.
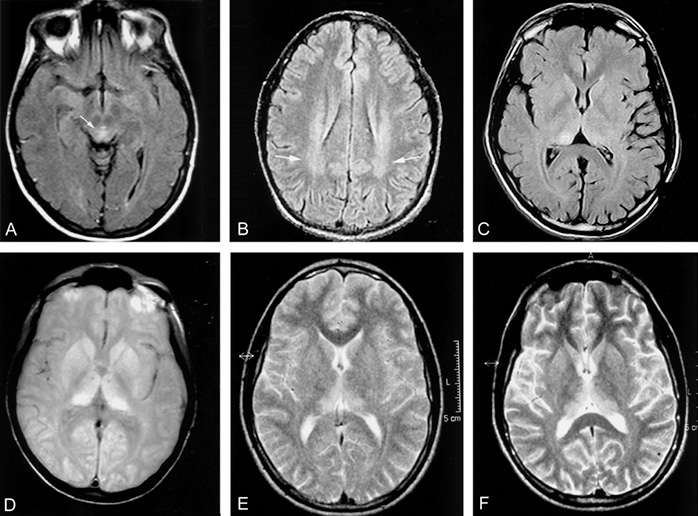
Первые симптомы сБКЯ обычно неспецифические: головная боль, недомогание, кашель, головокружение и изменение поведения, настроения или провалы в памяти. Для подтверждения диагноза должно пройти время, чтобы появились и иные основания полагать прионную природу. Классическими клиническими признаками с БКЯ являются:
- быстрое снижение когнитивных способностей;
- атаксия (нарушение согласованности движения различных мышц);
- миоклонус (быстрые внезапные сокращения отдельных мышц), оканчивающиеся акинетическим мутизмом (торможение всех двигательных функций, кроме фиксирующих движений глазных яблок).
Окончательный диагноз зависит от оценки клинических проявлений и результатов лабораторных тестов.
Акинетический мутизм — состояние при котором пациенты перестают двигаться и следить глазами за целью, за исключением реакции глаз на раздражители или длительной фиксации взгляда, их мышцы самостоятельно или под воздействием внешних факторов периодически быстро сокращаются. Пациенты страдают недержанием, не издают никаких звуков или только нечленораздельные шумы. Если глотание сохраняется, пациенты могут прожить в этом состоянии в течение нескольких недель, даже годы при иных благоприятных факторов, получая питание внутривенно или через трубку. При спорадической форме БКЯ, пациенты доходят до этого состояния в течение первых недель заболевания. Всамых стремительных сценариях, 10% пациентов доходят до этого состояния за год.
Долгое время самым информативным способом постановки диагноза было проведение диффузионно-тензорной МРТ. Этот способ является наиболее доступным, относительно неинвазивным и действенен при ранних изменениях в коре головного мозга. СБКЯ можно обнаружить через маркеры в назальных слизистых оболочках, спинномозговой жидкости, моче или крови, но эти тесты часто дают ложноположительные результаты — белок 14.3.3 не является специфичным без появления сопутствующей клинической картины. Протеинограмма является новым оптимальным методом диагностирования сБКЯ, так как она самая чувствительная из всех вышеназванных.
Белки 14-3-3 — семейство регуляторных молекул, встречающихся у всех эукариот. Они связываются со множеством других белков, регулируя их функции и тем самым влияя на множество процессов, в том числе регулировку клеточного цикла, контроль метаболизма, апоптоз, контроль транскрипции генов. Они были обнаружены более 40 лет назад при систематической классификации белков нервной ткани, где их содержание превышает 1% от всех белков. К настоящему времени описано более 300 различных белков-мишеней, способных взаимодействовать с 14-3-3.
Протеинограмма — исследование, изучающее количественное соотношение разновидностей белка в крови. В понятие общего белка входят все возможные белки, несмотря на их различия в строении и функциях.
Наследственная форма болезни Крейтцфельда – Якоба и также связанная с генетическими мутациями фатальная семейная бессонница составляют всего 10% от всех случаев прионных болезней. Множество исследований указывает на то, что общий путь в патогенезе заболевания может быть общим как для спорадических, так и для наследственных форм прионного заболевания, за исключением того, что в первом случае превращение белка происходит без участия каких-либо факторов, а не предопределено наличием мутации в генах.
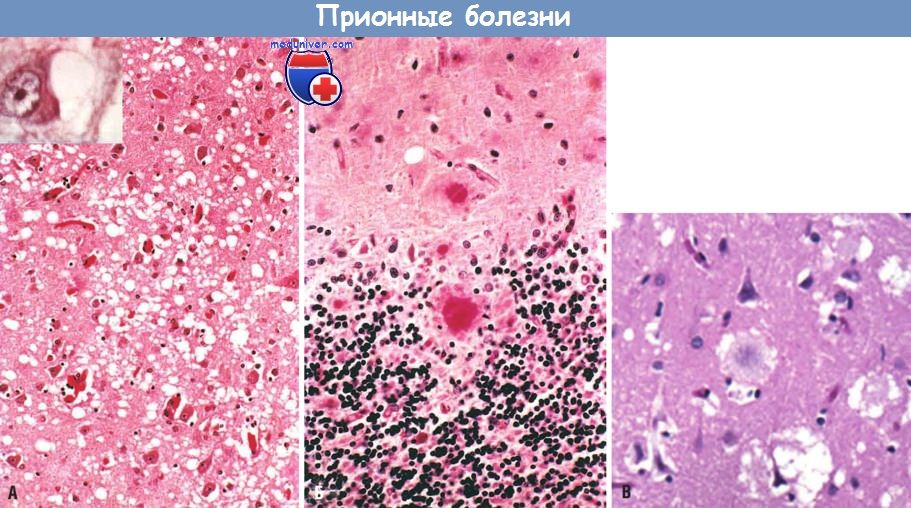
Фенотип — совокупность характеристик, присущих на определённой стадии развития болезни. Фенотип формируется на основе генотипа.
Полиморфизм — способность некоторых организмов существовать в состояниях с различной внутренней структурой или в разных внешних формах.
При наследственном прионном заболевании, фенотип заболевания будет определяться комбинированным эффектом патогенных мутаций, полиморфизма кодонов 129 и типа PrPSc. Полиморфизм кодона 129 играет двойную роль в прогнозировании исхода заболевания. Главным в понимании патогенеза прионной болезни является детальное и точное знание процессов и условий in vivo для образования PrPSc, которые неизбежно приводят к развитию и выражению заболевания. Эти знания позволят разработать рациональную и эффективную стратегию терапевтического вмешательства.
Новый вариант болезни Крейтцфельда – Якоба (nvCJD) был впервые идентифицирован в 1996 году. Последующие исследования подтвердили гипотезу, что эта форма связана с бычьей губчатой энцефалопатией. Скорее всего, пациенты употребляли в пищу мясо, содержащее патологические прионы мозга коров.
В отличии от спорадической формы, болезнь не имеет четкого возраста заражения. У пациентов с нБКЯ часто выявляют и психиатрические симптомы, потому порой она ошибочно диагностируется как психическое, а не неврологическое расстройство. Истинная причина психиатрических симптомов кроется в когнитивных нарушениях, постоянных болях в конечностях, нарушениях адекватности ощущений (парестезия или дизестезия), расстройствах речи или зрения.
В течение 6–8 месяцев развиваются пороки управления мышечной системой, но в некоторых случаях развитие болезни может длиться и более 18 месяцев. Потому этот диагноз достаточно трудно поставить при появлении первых признаков заболевания. Если у пациента возникают неконтролируемые движения, возрастает вероятность грамотного диагностирования нБКЯ. В отличие от спорадической формы, где характерны внезапные мышечные спазмы при напряжении (миоклонус), в случае с новой формой возможны и дистония (синдром, при котором происходит постоянное спазматическое сокращение мышц), и хорея (синдром, характеризующийся беспорядочными, отрывистыми, нерегулярными движениями).
Летальная стадия новой формы похожа на летальную стадию спорадической формы болезни Крейтцфельда-Якоба, она проходит с прогрессирующей потерей контроля над мышцами, часто приводящей к состоянию акинетического мутизма.
В отличие от более распространенных слабоумных состояний, которые обычно развиваются годами, быстро прогрессирующие деменции могут развиваться в течение нескольких месяцев, недель или даже дней и приводить к смерти. На спорадическую форму БКЯ приходится 46,9 % всех зарегистрированных случаев быстро прогрессирующей деменции, на генетическую форму прионных заболеваний — 13,6%. 39% всех случаев составляют лобно-височная деменция (FTD), кортикобазальная дегенерация (CBD), болезнь Альцгеймера (AD), деменция с тельцами Леви (DLB) и прогрессивный паралич.
Как правило, спорадическая форма БКЯ представлена совокупностью деменции и нейродегенеративных или психиатрические симптомов. У таких больных распространены пирамидная, мозжечковая и фокальная кортикальная дисфункция. У трети пациентов деменции предшествуют жалобы на усталость, головную боль, нарушение сна, недомогание, потерю веса, боль, депрессию или изменения в поведении.
Неврологические симптомы, включая атаксию, дизестезию, слабоумие или мышечные расстройства (хорея, миоклонус или дистония) появляются позже. Большинство случаев быстро прогрессирующей деменции без других сопутствующих симптомов случается у пожилых людей из-за метаболических нарушений или острых инфекций (пневмонии или инфекции мочевыводящих путей). Потому перед прохождением лабораторных тестов врачи первостепенно указывают быстро прогрессирующую деменцию без явного диагноза. Окончательный вердикт будет зависеть от полученных результатов анализов и обследований, заболевания будут отличаться в зависимости от клинической картины. ЭЭГ может помочь исключить судорожную активность головного мозга и обратиться к диагностике других состояний, таких как БКЯ.
Быстро прогрессирующая деменция представляют собой одну из самых сложных неврологических проблем. Дифференциальная диагностика широко используется для подтверждения окончательных диагнозов, которые могут относиться к нейродегенеративным, аутоиммунным, инфекционным и опухолевым заболеваниям. Даже при таком тщательном подходе к обследованию пациентов, небольшой процент случаев диагностируются уже после смерти.
Фатальная семейная бессонница — это редкое прионное заболевание, которое в буквальном смысле лишает сна и приводит к снижению всех нейро-двигательных и психических функций. Можно выделить две формы этой болезни: генетическую и спорадическую.
Генетическая форма связана с мутацией, приводящей к превращению белка PrP в прионный белок. Спорадическая же появляется спонтанно, без каких-либо предпосылок. Это заболевание отличается от других прионных заболеваний областью поражения — превращение прионных белков в патологические преимущественно происходит в одном отделе головного мозга — таламусе, который, в том числе, отвечает и за сон.
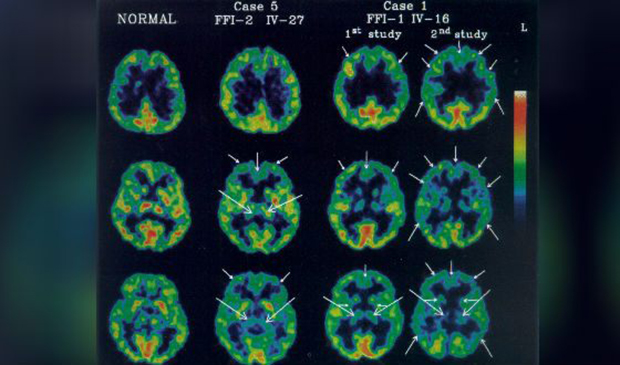
Диагноз фатальной семейной бессонницы в генетической форме подтверждается генетическим тестированием. В случае со спорадическими случаями, могут обнаружить нарушения в структуре сна и аномалии в таламусе полисомнография и позитронно-эмиссионная томография (ПЭТ). Средняя продолжительность жизни с начала первых симптомов заболевания — 3 года, лечения не существует.
Синдром Герстманна-Штраусслера-Шейнкера — это редкая генетическая форма трансмиссивной губчатой энцефалопатии, которая впервые была описана австрийскими неврологами в 1936 году. Синдром чаще всего проявляется в возрасте 40–50 лет, и вызван мутациями генов прионного белка PRNP на 20 хромосоме.
Клиническая картина синдром схожа со спорадической формой БКЯ, но он отличается продолжительностью и медленно прогрессирующей деменцией наряду с иными симптомами. Синдром Герстманна-Штраусслера-Шейнкера может длиться как нескольких месяцев так и несколько лет, средняя продолжительность жизни — 5 лет. Диагностировать заболевание можно даже на ранних стадиях посредством проведения магнитно-резонансной томографии. На МРТ будут наблюдаться губчатые изменения в коре и разрастание глиальных клеток.
Глиальные клетки или нейроглия — совокупность вспомогательных клеток нервной ткани. Составляет около 40 % объёма ЦНС. Количество глиальных клеток в мозге примерно равно количеству нейронов.
Глиальные клетки имеют общие функции и, частично, происхождение (исключение — микроглия). Они составляют специфическое микроокружение для нейронов, обеспечивая условия для генерации и передачи нервных импульсов, а также осуществляя часть метаболических процессов самого нейрона.
Обнаруживать прионную болезнь достаточно трудозатратно, учитывая то, что если диагноз подтвержден, пациенту уже ничем не помочь, а больницы теряют миллионы рублей. И самое жуткое, что все люди на Земле в группе риска. Прионы не пощадят никого. Поэтому не пробуйте фарш, после того, как его посолили. Не ешьте потроха и мозги коров и свиней, и посещайте врача вовремя. В конце концов, МРТ врать не будет.
Читайте также:
