
Что такое болезнь крейтцфельдта-якоба
(Подострая губчатая энцефалопатия)
, MD, Case Western Reserve University
Last full review/revision December 2018 by Pierluigi Gambetti, MD
БКЯ имеет три формы (1):
сБКЯ является наиболее распространенным типом, на который приходится около 85% случаев. сБКЯ обычно проявляется у пациентов в возрасте >40 лет (медиана – около 60 лет).
Семейная БКЯ диагностируется в около 5 до 15% случаев. Наследование аутосомно-доминантное, возраст начала заболевания, как правило, раньше, чем при сБКЯ с большей длительностью заболевания.
Приобретенная БКЯ вероятно, объясняет
Вариантная БКЯ (вБКЯ)
ВБКЯ редкая приобретенная форма БКЯ. Большинство случаев произошло в Великобритании (UK), которые составили 178 случаев в сентябре 2018 года, по сравнению с 53 случаями во всех других европейских и неевропейских странах. вБКЯ диагностировалась после приема в пищу мяса, полученного от крупного рогатого скота, заболевшего губкообразной энцефалопатией крупного рогатого скота (ГЭКРС), также называемой коровьим бешенством.
В случае заражения вБКЯ, симптомы развиваются в более раннем среднем возрасте ( 30 лет), чем в случае заражения сВКЯ. В недавно диагностированных случаях инкубационный период (время между употреблением в пищу зараженной говядины и развития симптомов) составлял от 12 до более чем 20 лет.
В начале 1980-х годов из-за слабо контролируемых правил переработки побочных продуктов животного происхождения, зараженной ткани, вероятно, от овец, зараженных скрепи, или крупного рогатого скота, зараженных ГЭКРС, скрепи прионового белка (PrP Sc ) попадали в корм для крупного рогатого скота. У сотен тысяч голов крупного рогатого скота развилась ГЭКРС. Несмотря на широкое воздействия, относительно у немногих людей, которые употребляли в пищу мясо больного крупного рогатого скота развилась вБКЯ.
В связи с длительным инкубационным периодом ГЭКРС связь между заболеванием и зараженным мясом в Великобритании не была установлена до тех пор, пока заболеваемость ГЭКРС не переросла в эпидемию. Эпидемия ГЭКРС перешла под контроль после массивного убоя скота и после изменения в процедурах производства технических фабрикатов, которые резко сократили загрязнения мяса тканями нервной системы. В Великобритании ежегодное число новых случаев вБКЯ, которое достигло пика в 2000 году, неуклонно сокращалось, и было зафиксировано только 2 случая после 2011 года.
Четыре случая вБКЯ были связаны с переливанием крови, они диагностировались у людей, которым была проведена трансфузия между 1996 и 1999. В Великобритании приблизительно 1 из 2000 человек могут быть носителями вБКЯ (на основании изучения большого количества образцов ткани аппендикса), но не имеют никаких симптомов; эти люди могут передавать болезнь, являясь донорами крови или при проведении хирургической процедуры. Таким образом, неясно, имеется ли группа пациентов, которым переливали инфицированную кровь и которые, таким образом, находятся в зоне риска последующего развития вБКЯ. Тем не менее, новые направления в критериях, связанных с донорством, связанные с вБКЯ, могут дополнительно снизить риск передачи вБКЯ при переливании крови, который уже достаточно низкий за пределами Франции и Великобритании.
Хотя нет официальных данных ни об одном случае возникновения вБКЯ в Северной Америке, были опубликованы сообщения о случаях ГЭКРС у нескольких особей крупного рогатого скота в Северной Америке (4 в США и 19 в Канаде).
Общие справочные материалы
2. Ritchie DL, Barria MA, Peden, AH, et al: UK Iatrogenic Creutzfeldt-Jakob disease: Investigating human prion transmission across genotypic barriers using human tissue-based and molecular approaches. Acta Neuropathol 133 (4): 579–595, 2017. doi: 10.1007/s00401-016-1638-x.
3. Cali I, Cohen ML, Haik S, et al: Iatrogenic Creutzfeldt-Jakob disease with amyloid-β pathology: An international study. Acta Neuropathol Commun 6 (1):5, 2018. doi: 10.1186/s40478-017-0503-z.
Клинические проявления
Приблизительно у 70% пациентов, заболевших БКЯ, выявляются нарушение памяти, внимания и изменение поведения, которые в конечном счете развиваются у всех пациентов; у 15–20% отмечается расстройство координации и атаксия, которые часто появляются на ранних стадиях заболевания. На более поздних стадиях могут возникнуть миоклонические судороги, вызываемые громким звуком (миоклония при испуге) или другими сенсорными стимулами. У людей с вБКЯ диагностируются психические симптомы (например, тревога, депрессия), а не потеря памяти. Более поздние симптомы похожи при обеих формах.
Помимо наиболее характерных для БКЯ деменции, атаксии и миоклонических судорог могут появиться и другие неврологические расстройства (например, галлюцинации, эпилептиформные припадки, нейропатия, различные двигательные нарушения).
При сБКЯ часто встречаются зрительные нарушения (например, дефекты поля зрения, диплопия, затуманенность или нечеткость зрения, зрительная агнозия).
Описание и причины патологии
Во время исследований анамнеза заразившихся пациентов было установлено, что заболевание передается через инфицирование во время оперативного вмешательства, пересадку биологических волокон от человека к человеку, переливание продуктов крови, неправильное применение гормональных препаратов. Если употреблять в пищу мясо зараженных домашних животных, также может произойти инфицирование.
Прион не относится к биологическому патогену или к вирусному штамму. Он представляет собой агрессивный белок, который в некотором количестве содержится в клеточном строении клеток головного мозга, но имеет несколько измененную структуру. Патогенное белковое соединение, проникая в тело человека, разносится по организму с током крови, не разрушается, а откладывается на нейронах. Здоровая белковая часть клетки, вступая во взаимодействие с агрессором, преобразуется в подобную структуру, становясь патогеном. С накоплением в клетке таких форм происходит образование внешней нейронной бляшки, которая постепенно умерщвляет клеточную структуру органа.
Первые феноменальные проявления в организме происходят спустя долгий инкубационный период. Это связано с естественной длительностью распространения аномального элемента с током крови до мозговых структур, встраиванием в клеточный состав и преобразованием нормальных белков в измененную форму. В зависимости от способа инфицирования, длительность инкубации варьируется от одного года до 12-13 лет. Так, если ткани мозга были заражены через медицинские инструменты во время операции, первые изменения в мозге проявляются примерно через 14-20 месяцев. Если прион попал в организм с трансплантированными волокнами, первичная симптоматика будет выявлена через пять – шесть лет, а при внутримышечном введении гормональных средств, выделенных из секреции крупного рогатого скота, инкубационный этап может затянуться до 12-13 лет. Еще реже имеют место случаи болезни генетического характера, приводящие к стимуляции выработки собственного трансформированного белка.
Явные признаки заболевания
Чаще всего развитие недуга носит затяжной, плавный характер, но отмечаются и пациенты, страдающие стремительной вариацией – острой формой, когда болезнь начинается с резких поведенческих изменений и поражения когнитивных функций. Треть заболевших жалуются на такие состояния, как:
- болевой синдром в области головы;
- частые предобморочные ощущения;
- раздражительность, подавленное настроение;
- рассеянность внимания;
- прерывистость ночного сна, бессонница;
- снижение уровня памяти;
- частичная потеря звуковосприятия;
- апатия к происходящему вокруг, потеря активности;
- отсутствие сексуального влечения;
- трансформация поведенческих реакций;
- иногда возникает беспричинная эйфория или паническая атака, бред или галлюцинации;
- частичная потеря координации;
- забывание слов, невозможность выполнить простейшие математические вычисления в уме.

При дальнейшем течении болезни происходит развитие последующей дегенеративной симптоматики:
- периодическая парализация мышечного аппарата;
- эпилептические припадки;
- сильная дрожь в конечностях;
- нервные подергивания отдельных мышечных групп, чаще всего носогубного треугольника и глазных век;
- прогрессирующее слабоумие, потеря мыслительных способностей;
- речевая дисфункция вплоть до полного распада осознанного звуковоспроизведения;
- у пациента пропадает чувствительность к внешнему воздействию.
На завершающей стадии недуга наблюдается:
- глубинная деменция;
- полная отрешенность, отсутствие контактности;
- потеря контроля функций выделения, больному требуется постоянный присмотр и уход;
- атрофия мышечного аппарата, дегенерация безусловных рефлексов (например, глотания);
- впадение в коматозное состояние с последующим летальным исходом.
Диагностические меры
Основываясь на прогрессирующей симптоматике, лечащий врач – невролог обращается к инструментальным методикам диагностирования: ЭЭГ, ПЭТ, магнитно-резонансное сканирование мозга головы, пункцию мозговых тканей. При затруднениях в диагностировании может быть использована проникающая биопсия. Это самый информативный способ диагностирования при данном недуге, так как у специалистов появляется возможность выявить повышенное количество патогенного приона в изъятых на анализ тканях.
Лечение аномального состояния
Восстановление пораженных волокон в современной медицине в настоящее время недоступно. Медики могут назначить только симптоматическое лечение, направленное на купирование психических проявлений, противоэпилептические препараты, медикаменты, назначаемые при болезни Паркинсона. Лечение известными противовирусными веществами не достигает какого-либо эффекта, также как и предварительная вакцинация пациентов и домашнего скота. Некоторый эффект выявлен при применении Брефелдина А, который купирует разрастание агрессивных белков в пораженных нейронах, но данный эффект является временным. Полностью избавиться от аномалии еще не удалось, но подходы к лечению находятся в стадии активной разработки.
Прогноз и профилактика
Так как медицинскими способами невозможно купировать проявления недуга, а трансформация приона происходит с высокой скоростью, единственным финалом для заболевших пациентов, к сожалению, является летальный исход. Средняя продолжительность жизни после проявления первых признаков составляет 8-12 месяцев в 90% случаев. Лишь небольшая часть больных продолжают жить в течение 24-26 месяцев.
Профилактические меры заключаются в тщательной обработке медицинских инструментов перед оперативным вмешательством, анализе тканей, предназначенных для целей трансплантологии и отказе от мясной продукции от крупного рогатого скота. Самые частые случаи заболеваемости описанной патологией фиксируются в таких странах, как Великобритания, Чили, Словакия и Израиль.
С появлением Интернета и свободных СМИ, люди стали узнавать всё больше о смертельных заболеваниях — инфекционных, вирусных, онкологических и наследственных. Но мало кто слышал о фатальных прионных болезнях. Несмотря на клинические испытания, на данный момент не существует ни одного доказанного универсального лечения этой группы заболеваний. Невролог Ричард Джонсон из Университета Джона Хопкинса говорит, что если прионы пациента превратились в патологические, он умирает, и мы не можем этого избежать.
Прионные заболевания — они же называются трансмиссивными губчатыми энцефалопатиями — представляют собой семейство редких прогрессирующих нейродегенеративных заболеваний, которые поражают как людей, так и животных. Их отличают:
- длительный инкубационный период;
- характерные губчатые разрыхления мозговой ткани, связанные с потерей нейронов;
- неспособность иммунной системы отреагировать на заражение, инициируя воспалительный процесс.
Прионные болезни поражают как людей, так и животных, быстро прогрессируют и всегда приводят к летальному исходу.
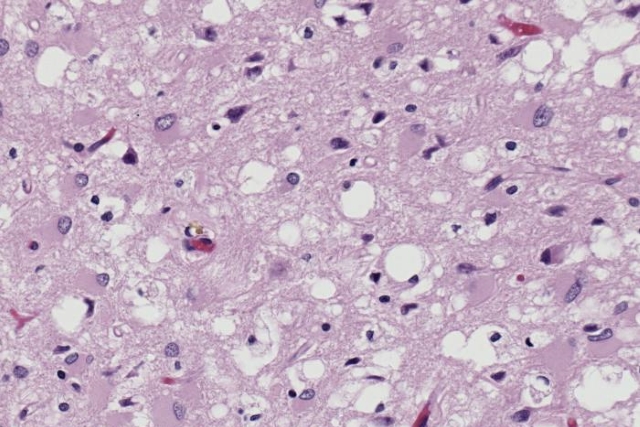
Возбудитель болезни — прионы — тип белков с аномальной третичной структурой, не содержащий нуклеиновых кислот. Сам термин относится к патологическим патогенным агентам, которые способны вызывать аномальное сворачивание специфических нормальных клеточных белков, которые называются как раз прионными белками, встречающихся чаще всего в мозге. Функции этих нормальных прионных белков до сих пор полностью не изучены.
Болезнь Крейтцфельдта – Якоба — БКЯ, псевдосклероз спастический, синдром кортико-стриоспинальной дегенерации, трансмиссивная спонгиоформная энцефалопатия, коровье бешенство.
Это прогрессирующее дистрофическое заболевание коры большого мозга, базальных ганглиев и спинного мозга. Считается основным проявлением губчатой энцефалопатии (прионной болезни). Излечение невозможно. БКЯ поражает людей всех национальностей и рас, мужчин и женщин, взрослых и детей.
Прионные белки — нормальные белки, которые присутствуют у каждого человека. Но есть определённые группы людей, у которых присутствует генетическая мутация, предрасполагающая их к синтезу патогенного прионного белка. Прионные болезни также могут передаваться путём прямого заражения, передача может произойти и в ходе хирургических манипуляций, использования человеческого гормона роста или употреблении заражённого мяса. Такой вид заражения называется ятрогенным и он остаётся в процентном меньшинстве относительно иных форм БКЯ.
Процентное соотношение ятрогенных случаев болезни Крейтцфельдта – Якоба в исследовании National CJD Research & Surveillance Unit у 177 пациентов.
- Гормон роста (соматотропин) — 53,1% (94 случая).
- Твердая мозговая оболочка (в том числе поедание) — 38,9% (69 случаев).
- Гонадотропный гормон — 2,25% (4 случая).
- Нейрохирургический инструментарий — 2,25% (4 случая).
- Пересадка роговицы — 1,69% (3 случая).
- Электроды для стереоэлектроэнцефалографии — 1,12% (2 случая).
- Пересадка печени — 0,56% (1 случай).
Есть случаи заражения, которые не классифицируются ни по одной из двух вышеупомянутых причин, в таком случае они считаются спорадическими, то есть возникшими спонтанно и самостийно, по независящим от генетики или внешних факторов обстоятельствам.
Доктор Ойбек Тургунхужаев, руководитель направления нейрореабилитации Междисциплинарного центра реабилитации (Москва), говорит, что окончательный диагноз человеку с подозрением на какое-либо прионное заболевание основывается на оценке клинических признаков и симптомов и ряде вспомогательных исследований. Долгое время единственным методом подтверждения диагноза была электроэнцефалография. Но поскольку общая чувствительность этого метода ограничена, полезность этого исследования была поставлена под сомнение.
Прионные заболевания неизлечимы, они неизбежно фатальны. Кроме этого, проблема заключается в том, что для постановки достоверного диагноза необходимо проводить вскрытие. Любое вскрытие — это риск для патологоанатома, так как были случаи ятрогенного заражения специалистов от умерших пациентов.
По приказу Роспотребнадзора, о том, что человек заболел прионной болезнью необходимо извещать в течение двух часов. При этом установление такого диагноза ведет за собой, по российским инструкциям, утилизацию всего оборудования, с которым пациент был в контакте. Именно поэтому, когда Медуза рассказывала случай одной из больных БКЯ, все клиники говорили о том, что у них нет оборудования для наблюдениях таких пациентов. На самом деле — это просто способ не потерять миллионы рублей, утилизируя даже аппарат МРТ. Если бы речь шла о сотнях поставленных диагнозов прионной болезни (например, в США регистрируется 300 случаев ежегодно, возможно, их больше), тогда речь шла бы о потери миллиардов рублей для российских больниц и бюджета. Именно поэтому официально диагноз не ставится, врачи не хотят об этом говорить, так как никакого официального распоряжения не существует, что диагноз ставить нельзя. В итоге выходит, что заболевание есть, смерти есть, а причины для родственников и умирающих людей — нет.
Им никто не скажет, что скорее всего родственники уже заразились. Никто не скажет, что нельзя пробовать сырой фарш или есть сырое мясо, тем более мозги. Также как из-за того, что диагноз не ставится, можно случайно пересадить орган больного прионной болезнью, тем самым заразив другого человека. Также это может произойти через хирургический инструмент (такие случае были, об этом ниже).
Когда мы просили хоть кого-то рассказать нам о прионных болезнях, практически никто не готов был говорить открыто. Так мы анонимно поговорили с врачом-неврологом одной из крупнейших московских больниц. «С прионами две проблемы. Во-первых, для постановки достоверного диагноза необходимо проводить вскрытие. Хотя формально (по российским руководствам, например) проводить вскрытие можно, хотя и в особенных условиях. Любое вскрытие — это, естественно, дополнительный риск для патологоанатомов, потому что были описаны случаи заражения патологоанатомов от умерших пациентов. никто не хочет переводить на них риск.
Во-вторых, так как прионные инфекции — это тяжело протекающие, неизлечимые заболевания (хотя и с довольно сложным путем передачи), в нашей стране чертовски сложные законы для регистрации и ведения таких пациентов; о случаях обязаны сообщать в случае выявления чуть ли не в течение двух часов, после постановки диагноза необходимо проводить уничтожение части в том числе дорогостоящего оборудования, которое, как может оказаться по факту, даже рядом не лежало с пациентом, нужно переоформлять документы и так далее.
Болезнь Крейтцфельда – Якоба (БКЯ) является одной из разновидностей прионных болезней. Это быстро прогрессирующее, фатальное нейродегенеративное заболевание, которое, как полагают, вызвано аномальной изоформой прионного белка. БКЯ встречается во всем мире, и согласно статистике, во всем мире заболевает 1 из миллиона человек.
Прионные болезни не идут по одному и тому же сценарию, у людей, страдающих одним и тем же прионным поражением могут разниться эпидемиология и патогенез. Болезнь Крейтцфельда-Якоба делят на несколько типов.
Спорадическая Болезнь Крейтцфельда-Якоба (сБКЯ) — наиболее распространенный вид трансмиссивных губчатых энцефалопатий человека, на долю которого приходится около 85% случаев зарегистрированных заболеваний прионной природы. СБКЯ имеет очень быстрое течение болезни — средняя продолжительность жизни после проявления признаков составляет всего шесть месяцев. Более 90% пациентов умирают в течение года после появления симптомов. Пик заболеваемости приходится на пожилых людей возрастом 60–70 лет, в других возрастных группах случается куда реже. Одной из гипотез происхождения сБКЯ является мнение, что это спонтанное нейродегенеративное заболевание, возникающее в результате соматической мутации гена PRNP или случайного структурного изменения в белке PrP, вызывающее образование PrPSc2. Эпидемиологические исследования не выявили связи спорадической формы БКЯ с экологическими факторами.
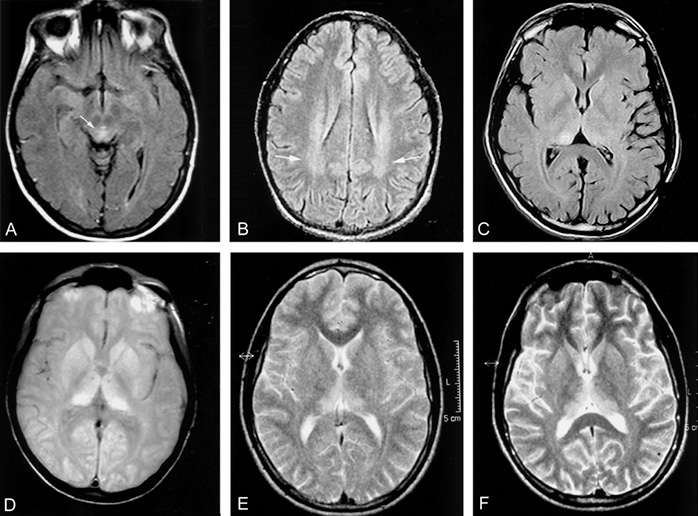
Первые симптомы сБКЯ обычно неспецифические: головная боль, недомогание, кашель, головокружение и изменение поведения, настроения или провалы в памяти. Для подтверждения диагноза должно пройти время, чтобы появились и иные основания полагать прионную природу. Классическими клиническими признаками с БКЯ являются:
- быстрое снижение когнитивных способностей;
- атаксия (нарушение согласованности движения различных мышц);
- миоклонус (быстрые внезапные сокращения отдельных мышц), оканчивающиеся акинетическим мутизмом (торможение всех двигательных функций, кроме фиксирующих движений глазных яблок).
Окончательный диагноз зависит от оценки клинических проявлений и результатов лабораторных тестов.
Акинетический мутизм — состояние при котором пациенты перестают двигаться и следить глазами за целью, за исключением реакции глаз на раздражители или длительной фиксации взгляда, их мышцы самостоятельно или под воздействием внешних факторов периодически быстро сокращаются. Пациенты страдают недержанием, не издают никаких звуков или только нечленораздельные шумы. Если глотание сохраняется, пациенты могут прожить в этом состоянии в течение нескольких недель, даже годы при иных благоприятных факторов, получая питание внутривенно или через трубку. При спорадической форме БКЯ, пациенты доходят до этого состояния в течение первых недель заболевания. Всамых стремительных сценариях, 10% пациентов доходят до этого состояния за год.
Долгое время самым информативным способом постановки диагноза было проведение диффузионно-тензорной МРТ. Этот способ является наиболее доступным, относительно неинвазивным и действенен при ранних изменениях в коре головного мозга. СБКЯ можно обнаружить через маркеры в назальных слизистых оболочках, спинномозговой жидкости, моче или крови, но эти тесты часто дают ложноположительные результаты — белок 14.3.3 не является специфичным без появления сопутствующей клинической картины. Протеинограмма является новым оптимальным методом диагностирования сБКЯ, так как она самая чувствительная из всех вышеназванных.
Белки 14-3-3 — семейство регуляторных молекул, встречающихся у всех эукариот. Они связываются со множеством других белков, регулируя их функции и тем самым влияя на множество процессов, в том числе регулировку клеточного цикла, контроль метаболизма, апоптоз, контроль транскрипции генов. Они были обнаружены более 40 лет назад при систематической классификации белков нервной ткани, где их содержание превышает 1% от всех белков. К настоящему времени описано более 300 различных белков-мишеней, способных взаимодействовать с 14-3-3.
Протеинограмма — исследование, изучающее количественное соотношение разновидностей белка в крови. В понятие общего белка входят все возможные белки, несмотря на их различия в строении и функциях.
Наследственная форма болезни Крейтцфельда – Якоба и также связанная с генетическими мутациями фатальная семейная бессонница составляют всего 10% от всех случаев прионных болезней. Множество исследований указывает на то, что общий путь в патогенезе заболевания может быть общим как для спорадических, так и для наследственных форм прионного заболевания, за исключением того, что в первом случае превращение белка происходит без участия каких-либо факторов, а не предопределено наличием мутации в генах.
Фенотип — совокупность характеристик, присущих на определённой стадии развития болезни. Фенотип формируется на основе генотипа.
Полиморфизм — способность некоторых организмов существовать в состояниях с различной внутренней структурой или в разных внешних формах.
При наследственном прионном заболевании, фенотип заболевания будет определяться комбинированным эффектом патогенных мутаций, полиморфизма кодонов 129 и типа PrPSc. Полиморфизм кодона 129 играет двойную роль в прогнозировании исхода заболевания. Главным в понимании патогенеза прионной болезни является детальное и точное знание процессов и условий in vivo для образования PrPSc, которые неизбежно приводят к развитию и выражению заболевания. Эти знания позволят разработать рациональную и эффективную стратегию терапевтического вмешательства.
Новый вариант болезни Крейтцфельда – Якоба (nvCJD) был впервые идентифицирован в 1996 году. Последующие исследования подтвердили гипотезу, что эта форма связана с бычьей губчатой энцефалопатией. Скорее всего, пациенты употребляли в пищу мясо, содержащее патологические прионы мозга коров.
В отличии от спорадической формы, болезнь не имеет четкого возраста заражения. У пациентов с нБКЯ часто выявляют и психиатрические симптомы, потому порой она ошибочно диагностируется как психическое, а не неврологическое расстройство. Истинная причина психиатрических симптомов кроется в когнитивных нарушениях, постоянных болях в конечностях, нарушениях адекватности ощущений (парестезия или дизестезия), расстройствах речи или зрения.
В течение 6–8 месяцев развиваются пороки управления мышечной системой, но в некоторых случаях развитие болезни может длиться и более 18 месяцев. Потому этот диагноз достаточно трудно поставить при появлении первых признаков заболевания. Если у пациента возникают неконтролируемые движения, возрастает вероятность грамотного диагностирования нБКЯ. В отличие от спорадической формы, где характерны внезапные мышечные спазмы при напряжении (миоклонус), в случае с новой формой возможны и дистония (синдром, при котором происходит постоянное спазматическое сокращение мышц), и хорея (синдром, характеризующийся беспорядочными, отрывистыми, нерегулярными движениями).
Летальная стадия новой формы похожа на летальную стадию спорадической формы болезни Крейтцфельда-Якоба, она проходит с прогрессирующей потерей контроля над мышцами, часто приводящей к состоянию акинетического мутизма.
В отличие от более распространенных слабоумных состояний, которые обычно развиваются годами, быстро прогрессирующие деменции могут развиваться в течение нескольких месяцев, недель или даже дней и приводить к смерти. На спорадическую форму БКЯ приходится 46,9 % всех зарегистрированных случаев быстро прогрессирующей деменции, на генетическую форму прионных заболеваний — 13,6%. 39% всех случаев составляют лобно-височная деменция (FTD), кортикобазальная дегенерация (CBD), болезнь Альцгеймера (AD), деменция с тельцами Леви (DLB) и прогрессивный паралич.
Как правило, спорадическая форма БКЯ представлена совокупностью деменции и нейродегенеративных или психиатрические симптомов. У таких больных распространены пирамидная, мозжечковая и фокальная кортикальная дисфункция. У трети пациентов деменции предшествуют жалобы на усталость, головную боль, нарушение сна, недомогание, потерю веса, боль, депрессию или изменения в поведении.
Неврологические симптомы, включая атаксию, дизестезию, слабоумие или мышечные расстройства (хорея, миоклонус или дистония) появляются позже. Большинство случаев быстро прогрессирующей деменции без других сопутствующих симптомов случается у пожилых людей из-за метаболических нарушений или острых инфекций (пневмонии или инфекции мочевыводящих путей). Потому перед прохождением лабораторных тестов врачи первостепенно указывают быстро прогрессирующую деменцию без явного диагноза. Окончательный вердикт будет зависеть от полученных результатов анализов и обследований, заболевания будут отличаться в зависимости от клинической картины. ЭЭГ может помочь исключить судорожную активность головного мозга и обратиться к диагностике других состояний, таких как БКЯ.
Быстро прогрессирующая деменция представляют собой одну из самых сложных неврологических проблем. Дифференциальная диагностика широко используется для подтверждения окончательных диагнозов, которые могут относиться к нейродегенеративным, аутоиммунным, инфекционным и опухолевым заболеваниям. Даже при таком тщательном подходе к обследованию пациентов, небольшой процент случаев диагностируются уже после смерти.
Фатальная семейная бессонница — это редкое прионное заболевание, которое в буквальном смысле лишает сна и приводит к снижению всех нейро-двигательных и психических функций. Можно выделить две формы этой болезни: генетическую и спорадическую.
Генетическая форма связана с мутацией, приводящей к превращению белка PrP в прионный белок. Спорадическая же появляется спонтанно, без каких-либо предпосылок. Это заболевание отличается от других прионных заболеваний областью поражения — превращение прионных белков в патологические преимущественно происходит в одном отделе головного мозга — таламусе, который, в том числе, отвечает и за сон.
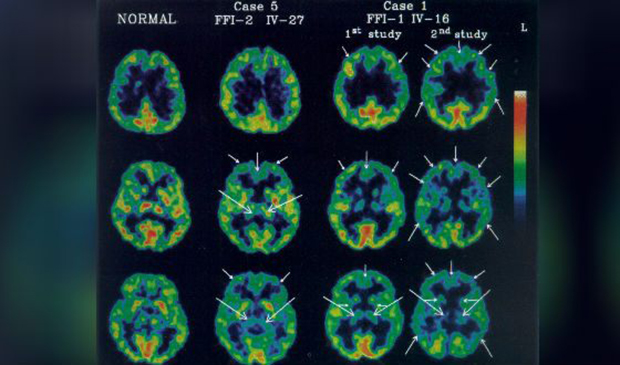
Диагноз фатальной семейной бессонницы в генетической форме подтверждается генетическим тестированием. В случае со спорадическими случаями, могут обнаружить нарушения в структуре сна и аномалии в таламусе полисомнография и позитронно-эмиссионная томография (ПЭТ). Средняя продолжительность жизни с начала первых симптомов заболевания — 3 года, лечения не существует.
Синдром Герстманна-Штраусслера-Шейнкера — это редкая генетическая форма трансмиссивной губчатой энцефалопатии, которая впервые была описана австрийскими неврологами в 1936 году. Синдром чаще всего проявляется в возрасте 40–50 лет, и вызван мутациями генов прионного белка PRNP на 20 хромосоме.
Клиническая картина синдром схожа со спорадической формой БКЯ, но он отличается продолжительностью и медленно прогрессирующей деменцией наряду с иными симптомами. Синдром Герстманна-Штраусслера-Шейнкера может длиться как нескольких месяцев так и несколько лет, средняя продолжительность жизни — 5 лет. Диагностировать заболевание можно даже на ранних стадиях посредством проведения магнитно-резонансной томографии. На МРТ будут наблюдаться губчатые изменения в коре и разрастание глиальных клеток.
Глиальные клетки или нейроглия — совокупность вспомогательных клеток нервной ткани. Составляет около 40 % объёма ЦНС. Количество глиальных клеток в мозге примерно равно количеству нейронов.
Глиальные клетки имеют общие функции и, частично, происхождение (исключение — микроглия). Они составляют специфическое микроокружение для нейронов, обеспечивая условия для генерации и передачи нервных импульсов, а также осуществляя часть метаболических процессов самого нейрона.
Обнаруживать прионную болезнь достаточно трудозатратно, учитывая то, что если диагноз подтвержден, пациенту уже ничем не помочь, а больницы теряют миллионы рублей. И самое жуткое, что все люди на Земле в группе риска. Прионы не пощадят никого. Поэтому не пробуйте фарш, после того, как его посолили. Не ешьте потроха и мозги коров и свиней, и посещайте врача вовремя. В конце концов, МРТ врать не будет.
Читайте также:
