
Вирусные иммунодефициты гепатит с
Болезненные состояния, связанные с недостаточностью иммунной системы. Классификация иммунодефицитных состояний:
1) первичные (врожденные) — очень редкие, причиной является генетически обусловленный дефект иммунной системы: нарушение образования антител (наиболее частый — общая вариабельная иммунная недостаточность [ОВИН]), нарушения клеточного ответа, нарушения фагоцитоза, дефицит комплемента и другие очень редкие нарушения;
2) вторичные (приобретенные) — вызванные действием внешних факторов или болезнью; чаще всего они имеют смешанный характер (нарушение специфического ответа [гуморального и клеточного] и неспецифического [напр. нарушения системы комплемента]); главные причины иммуносупрессивная терапия, инфекции (ВИЧ, вирус кори, HSV, бактериальные [в том числе микобактериальные] и паразитарные [малярия]), опухоль (CLL, лимфома Ходжкина, моноклональная гаммапатия, солидные опухоли), метаболические нарушения (при сахарном диабете, почечной недостаточности, печеночной недостаточности, недостаточном питании), аутоиммунные заболевания (СКВ, РА, синдром Фелти), ожоги, факторы окружающей среды (ионизирующее излучение, химические соединения), беременность, стресс, отсутствие селезенки (врожденная аспления или состояние после спленэктомии) или повреждение ее функции (функциональная аспления — гипоспленизм, в течении различных заболеваний с вовлечением селезенки), цирроз печени, старение; приобретенное снижение количества лейкоцитов в крови →разд. 27.1.
Частые, хронические и рекуррентные инфекции или (реже) симптомы аутоиммунизации. Инфекции имеют тяжелое, нередко нетипичное и длительное течение, устойчивы к антибиотикотерапии. Могут быть вызваны микроорганизмами, которые у здоровых людей редко вызывают инфекцию, напр., Mycobacterium avium, Cryptosporidium parvum , ЦМВ, Candida albicans . Характерными для дефицитов гуморального иммунитета являются рецидивирующие инфекции дыхательных путей и околоносовых пазух, вызванные инкапсулированными бактериями (напр., Haemophilus influenzae , Streptococcus pneumoniae ). Диагностику часто затрудняют ложноотрицательные серологические результаты исследований. Часто возникают аллергические реакции на антибиотики и пищевые аллергены.
Иммунодефицит следует подозревать у каждого пациента, с рецидивирующими или тяжелыми вирусными и/или бактериальными инфекциями, а также инфекциями, вызванными оппортунистическими возбудителями. Следует провести исследования, оценивающие отдельные компоненты иммунного ответа — сначала скрининговые, а затем более специализированные.
Сигнальные симптомы первичного иммунодефицита у взрослого человека:
1) 6 симптомов согласно ESID (2008): ≥4 инфекции, требующие применения антибиотикотерапии (отит, бронхит, синусит или пневмония) в течение года; рецидивирующие инфекции или инфекции, требующие долгосрочной антибиотикотерапии; ≥2 тяжелые бактериальные инфекции (остеомиелит, менингит, панникулит, сепсис); ≥2 случаев рентгенологически подтвержденных пневмоний в течение 3 лет; инфекции с нетипичной локализацией или вызванные нетипичными патогенами; наличие в семейном анамнезе первичного иммунодефицита;
2) 10 симптомов согласно Jeffrey Model Foundation (2013): ≥2 случаев отита в течение года; ≥2 случаев синусита в течение года у лица без аллергии; 1 случай пневмонии/год в течение >1 года; хроническая диарея с потерей массы тела; рецидивирующие вирусные инфекции (простуды, инфекция, вызванная вирусами Herpes, HPV); рецидивирующая необходимость использования внутривенной антибиотикотерапии; рецидивирующие глубокие абсцессы кожи или внутренних органов, персистирующие грибковые инфекции; инфекции, вызванные атипичными микобактериями; наличие в семейном анамнезе первичного иммунодефицита.
Оценка гуморального ответа
1 . Скрининговые исследования: концентрация иммуноглобулинов (IgG, IgM, IgA) в сыворотке, титр специфических антител (направленных против антигенов вакцин, введённых в детстве), оценка специфического титра антител в ответ на бустерную иммунизацию, определение количества В-клеток методом проточной цитометрии.
2 . Расширенные исследования: определение субпопуляции В-лимфоцитов методом проточной цитометрии, синтез иммуноглобулинов in vitro в ответ на митогены, CD40 и цитокины, оценка титров специфических антител в ответ на вакцинацию φX174.
Оценка клеточного иммунного ответа
1 . Скрининговые исследования: морфология периферической крови с мазком — оценка доли лимфоцитов и остальных видов лейкоцитов, определение количества (субпопуляций) Т-лимфоцитов и NK-клеток методом проточной цитометрии, кожные тесты исследование замедленной гиперчувствительности кожи (проба реакции на введение внутрикожного антигена; напр., БЦЖ, туберкулиновый тест), рентгенологические исследования тимуса, исследование способности спонтанной цитотоксичности NK-клеток.
2 . Расширенные исследования: определение субпопуляции Т-лимфоцитов методом проточной цитометрии, исследование цитотоксических способностей Т-лимфоцитов, ферментативные исследования (аденозиндезаминаза, пуриннуклеозидфосфорилаза), in vitro тест пролиферативного ответа на стимуляцию митогеном или антигеном, исследование in vitro синтеза и секреции цитокинов, а также и экспрессии поверхностных маркеров в ответ на стимуляцию митогеном или антигеном, анализ фосфорилирования цитоплазматических белков после стимуляции митогеном, исследование методом FISH о наличии делеции 22q11 и 10p11.
Оценка функции фагоцитарных клеток
1 . Скрининговые исследования: морфология периферической крови с мазком, оценка морфологии нейтрофилов при стандартном окрашивании, тест снижения NBT, оценка наличия адгезивных молекул методом проточной цитометрии.
2 . Расширенные исследования: хемилюминесценция (используется для оценки активности окислительных процессов в фагоцитарных клетках), исследования хемотаксиса и фагоцитоза методом проточной цитометрии, цитохимическое исследования — активность пероксидазы гранулоцитов, глюкозо-6-фосфатдегидрогеназы, тест способности уничтожения бактерий или грибков, биопсия костного мозга — количественная и морфологическая оценка клеток миелоидной линии.
Оценка системы комплемента
1 . Скрининговые исследования: исследование общей гемолитической активности комплемента (CH50), исследование гемолитической активности альтернативного пути активации комплемента (AH50).
2 . Расширенные исследования: концентрация или активность отдельных компонентов комплемента, хемотаксическая активность продуктов расщепления компонентов комплемента.
1 . Избегание ситуации, способствующих инфицированию.
2 . Устранение причины вторичного иммунодефицита.
3. Заместительная терапия внутривенными препаратами иммуноглобулинов (препараты ВВИГ →разд. 24.22.6.4): при иммунодефицитах, протекающих с гипо- или агаммаглобулинемией. Период полураспада IgG составляет ≈21 дней, поэтому рекомендуется введение ВВИГ каждые 21–28 дней, для получения защитной концентрации IgG (≥500 мг/дл). У больных с агаммаглобулинемией или тяжелой гипогаммаглобулинемией IgG ( 4. Профилактическая антибиотикотерапия: амоксициллин (500 мг/сут. или 250–500 мг 2 × в день) или котримоксазол (160 мг триметоприм 1 × в день или 80–160 мг 2 × в день) или азитромицин 500 мг 1 × в нед. Если эти лекарства окажутся неэффективными → кларитромицин 500 мг/сут. или амоксициллин с клавуланатом 875 мг или 1000 мг 1×в день. Показания: тяжелая или умеренная гипогаммаглобулинемия, если заместительная терапия IgG не препятствует частым инфекциям; тяжелый дефицит IgA или подклассов IgG, протекающий с частыми инфекциями; профилактика инфицирования Pneumocystis jirovecii , рекомендуется у больных с тяжелым комбинированным иммунодефицитом (ТКИД) и у больных, получающих интенсивную иммуносупрессивную терапию.
5 . Факторы роста G-CSF и GM-CSF: используются при нейтропениях. Они могут ускорить разрешение нейтропении различной этиологии (с тяжелой врожденной нейтропенией, циклической нейтропенией и СПИДом включительно), а также уменьшить тяжесть и сократить длительность инфекции. Во время проведения противоопухолевой терапии рассмотрите применение у больных с нейтропенией и плохим общим состоянием, в которых высокий риск инфекционных осложнений, особенно если раньше лечение рака осложнялось инфекциями или возникновением нейтропенической лихорадки. Препараты G-CSF: филграстим 3,45–11,5 мкг/кг/сут п/к, ленограстим, GM-CSF 300 мкг/сут.
6 . ИФН-α i IFN-γ: при врожденных дефицитах гуморального иммунитета (напр., ОВИН), при дефектах фагоцитарных клеток (напр., хроническая гранулематозная болезнь).
7 . алло-ТГСК: при некоторых первичных иммунодефицитах.
8 . Вакцинация больных с иммунодефицитом →разд. 18.11.
9 . Переливания клеточных компонентов крови: у пациентов с нарушением клеточного иммунитета проведите трансфузию только облученных (чтобы уменьшить количество лимфоцитов) концентратов эритроцитов или тромбоцитов от ЦМВ-отрицательных доноров.
10 . Алгоритм действий при нейтропенической лихорадке →разд. 22.2.5.
11. Тактика при асплении: в связи с высоким риском молниеносно протекающих бактериальных инфекций, характеризующихся высоким риском летального исхода, показано проведение вакцинаций против капсульных бактерий ( S. pneumoniae , H. influenzae типа b, N. meningitidis ), а также ежегодно против гриппа. В случае спленэктомии в плановом порядке, вакцинацию следует провести не позднее 2 нед. перед операцией, а если это невозможно, тогда через короткое время после операции. В случае появления лихорадки или озноба (предупреждающие симптомы) пациент с аспленией должен немедленно принять первую дозу антибиотика, который должен иметь при себе (амоксициллина с клавуланатом, цефуроксима, левофлоксацина или моксифлоксацина), и обратится к врачу; в последующем необходимо немедленно применить эмпирическую антибиотикотерапию широкого спектра (напр., цефтриаксон или цефотаксим в комбинации с ванкомицином).
5.8. ИММУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ
Иммунодефицитные состояния (ИДС) – это состояния, характеризую-щиеся снижением активности или неспособностью организма к эффективно-му осуществлению реакций клеточного и/или гуморального звена иммуните-та.
По происхождению все ИДС подразделяются на:
- первичные (наследственные, врожденные). Они являются результатом генетического дефекта, обусловливающего нарушения процессов про-лиферации, дифференцировки и функционирования клеток иммуноком-петентной системы;
- вторичные (приобретенные в постнатальном периоде). Развиваются под влиянием различных факторов физического или биологического харак-тера.
По преимущественному повреждению клеток иммунокомпетентной систе-мы различают 4 группы ИДС:
- с преимущественным повреждением клеточного иммунитета (Т-зависимые, клеточные);
- с преимущественным повреждением гуморального иммунитета (В-зависимые, гуморальные);
- с поражением системы фагоцитоза (А-зависимые);
- комбинированные, с поражением клеточного и гуморального звеньев иммунитета (Стефани Д.С.,ВельтищевЮ.Е., 1996; Ледванов М. Ю. , 1997; Резник И.Б.,1998; Хаитов Р.М.,Пинегин Б.В.,1999).
Физиологические ИДС включают в себя иммунодефициты (ИД) новорож-денных, беременных и лиц старческого возраста.
1. Иммунодефицит новорожденных. К моменту рождения у здоровых детей в крови содержатся материнские IgG и небольшое количество собст-венных IgG, IgM, IgA. Иммуноглобулины, полученные от матери, содержат антитела против всех видов микробов, с которыми контактировала мать, бла-годаря чему ребенок оказывается защищенным от них на протяжении первых месяцев жизни. Уровень материнских иммуноглобулинов постепенно снижа-ется, и максимальный дефицит их наблюдается через 2-3 месяца после рож-дения. Затем уровень собственных иммуноглобулинов ребенка в крови начи-нает постепенно повышаться, и количество IgM достигает нормального уровня взрослого человека в конце 1-го года жизни у мальчиков и 2-го – у девочек; IgG1 и IgG4 в возрасте 6-8 лет; IgG3 – в 10, а IgG2 - в 12 лет. Кон-центрация IgE достигает нормального уровня взрослого лишь спустя 10-15 лет после рождения. Секреторные IgA отсутствуют у новорожденных и по-являются через 3 месяца после рождения. Оптимальная концентрация секре-торного IgA устанавливается в возрасте 2-4 лет. Плазменный уровень IgA достигает такового показателя у взрослых в 10-12 лет. ИД новорожденных обусловлен тем, что высокое содержание лимфоцитов в периферической крови у новорожденных сочетается с их низкой активностью. У новорожден-ных детей отмечаются также низкая фагоцитарная активность и опсонизи-рующая способность крови. Уровень комплемента у новорожденных снижен и достигает уровня взрослого человека к 3-6-му месяцу жизни.
2. Иммунодефицит беременных. Иммунный статус беременных отлича-ется снижением числа Т- и В-лимфоцитов. Одновременно отмечается повы-шение активности С3-комплемента, что объясняют влиянием плацентарных стероидов на его синтез в печени.
3. Иммунодефицит лиц старческого возраста. Недостаточность имму-нитета при старении проявляется в снижении активности его гуморального и клеточного звеньев. При старении уменьшается общее число лимфоцитов пе-риферической крови. Функциональная активность Т- и В-лимфоцитов при старении падает, снижается интенсивность образования антител в ответ на антигенную стимуляцию. В старческом возрасте в основном продуцируются антитела класса IgM, резко снижена продукция IgA, IgG, подавляется синтез IgE, в связи с чем ослабевает течение атопических аллергических реакций. По мере старения уменьшается фагоцитарная активность макрофагов, ней-трофилов, снижаются активность комплемента, лизоцима и бактерицидная активность сыворотки крови.
Первичные ИДС - это генетически обусловленная неспособность орга-низма реализовать то или иное звено иммунного ответа. Эндогенные, как правило, генетически обусловленные дефекты одного из компонентов им-мунной системы приводят к нарушению системы защиты организма и кли-нически выявляются как одна из форм первичного ИДС. Так как в нормаль-ном функционировании иммунной системы и иммунном ответе участвуют многие типы клеток и сотни молекул, в основе первичного иммунодефицита лежат многочисленные варианты дефектов. Научная группа ВОЗ в 1997 г. выделила более 70 идентифицированных генетических дефектов на различ-ных уровнях преобразования стволовых клеток в Т- и В-лимфоциты или по-следующих этапах их дифференцировки, лежащих в основе первичных ИДС.
В последнее время в связи с обнаружением молекулярных дефектов, со-ставляющих основу многих иммунодефицитов, и существенной вариабель-ностью клинической картины и тяжести их течения, возможностью их позд-ней манифестации, в том числе у взрослых, становится ясно, что первичные ИДС – это не столь редкое состояние, как это считалось до сих пор. Частота значительной части первичных ИДС составляет 1/25000 - 1/50000, хотя такие варианты врожденных иммунных дефектов, как селективный дефицит IgA, встречается с частотой 1/500 - 1/700 человек. По данным ряда авторов, не-достаточность В-системы лимфоцитов и гуморального звена иммунитета от-мечается у 50-75% из общего числа больных ИДС; в 20% случаев отмечается комбинированная недостаточность клеточного и гуморального иммунитета; в 10% - изолированная недостаточность клеточного иммунитета, в 18% - не-достаточность фагоцитоза и в 2% - недостаточность системы комплемента.
ИДС с преимущественным нарушением клеточного звена иммунитета
Патология клеточного звена иммунитета проявляется на различных этапах созревания Т-лимфоцитов - от стволовой клетки до развития их специализи-рованных субпопуляций (рис. 1).
При дефектах преимущественно клеточного звена иммунитета характер-ны частые инфекции дыхательных и мочевыводящих путей, упорные рас-стройства пищеварения, хронический генерализованный кандидоз кожи и слизистых оболочек полости рта, пищеварительного тракта. Кандидозное по-ражение может выявляться в первые месяцы жизни в виде стоматита, дерма-тита, реактивной гиперплазии аденоидной ткани миндалин, лимфатических узлов, отмечается высокая интенсивность кариеса, развиваются бронхоле-гочная патология, фурункулез, в слюне повышено содержание секреторных IgA. Cледует отметить, что CD8 Т-лимфоциты осуществляют иммунологиче-ский надзор за внутренней средой, обеспечивая, в частности, элиминацию клеток, подвергшихся онкогенной трансформации. В случае недостаточности Т-системы лимфоцитов возникает онкогенноопасная ситуация.
1. Синдром Ди Джорджи возникает при гипо- и аплазии вилочковой же-лезы и паращитовидных желез. Заболевание обусловлено нарушением эм-бриональной дифференцировки эпителия в области 3-го и 4-го глоточных карманов. Число лимфоцитов в периферической крови значительно снижено. Синтез гуморальных антител не нарушен, но отмечается дефект в дифферен-цировке стволовых клеток в Т-лимфоциты. У детей с синдромом Ди Джорд-жи не отторгаются кожные трансплантаты, отсутствуют реакции ГЗТ. Забо-левание проявляется в периоде новорожденности (гипокальциемия, судороги, признаки кандидомикоза, инфекции дыхательных и мочевыводящих путей, упорные расстройства пищеварения).
2. Лимфоцитарная дисгенезия (синдром Незелофа) – количественная и качественная недостаточность Т-системы в результате атрофии тимуса и лимфатических узлов. Характеризуется отсутствием клеточных реакций им-мунологической защиты при нормальном содержании иммуноглобулинов в плазме крови. Проявляется в первые недели и месяцы жизни. Отмечаются за-держка развития ребенка, затяжной септический процесс с гнойно-воспалительными очагами во внутренних органах и коже. В периферической крови отмечается крайне низкое содержание лимфоцитов; резко угнетена ре-акция бласттрансформации лимфоцитов; слабо выражена реакция ГЗТ. Со-держание иммуноглобулинов всех классов в периферической крови – в пре-делах нормы. Дети чаще погибают в первые месяцы жизни от сепсиса.
ИДС с преимущественным повреждением В-системы
Гуморальные иммунодефициты относят к наиболее распространенным формам первичных ИДС.
2. Общая вариабельная гипогаммаглобулинемия (ОВГ). Это гетероген-ная группа ИДС, развитие которых связано с нарушением способности В-лимфоцитов трансформироваться в плазмоциты на фоне антигенной стиму-ляции. Количество циркулирующих в крови В-лимфоцитов не отличается от нормы, однако имеет место нарушение их дифференцировки. Для больных характерны гиперплазия лимфатических узлов, лимфоидного глоточного кольца, иногда – увеличение размеров селезенки. У детей с ОВГ не формиру-ется специфический иммунитет после вакцинации; у них обнаруживается склонность к развитию рецидивирующих воспалительных процессов инфек-ционной природы (синуситы, отиты, хронические пневмонии, фарингиты, тонзиллиты и др.). У взрослых больных ОВГ часто развиваются восходящий холангит и желчнокаменная болезнь, иногда артриты, атрофический гастрит. Общее количество иммуноглобулинов, особенно IgG, снижено.
Недостаточность местного иммунитета у больных ОВГ не коре-лирует с концентрацией плазматического уровня IgA и, вероятно, связана с наруше-ниями синтеза секреторных иммуноглобулинов. У многих больных отмечена склонность к аутоиммунным процессам. Установлено, что у больных ОВГ резко активированы естественные клетки-киллеры, причем их активность в 5 раз превышает нормальные значения.
3. Селективный дефицит иммуноглобулинов. Возможно развитие ИДС с селективным нарушением синтеза IgG, IgA. В основе их формирования могут лежать как блокада развития отдельных субпопуляций В-лимфоцитов, так и, что бывает чаще, повышение активности CD8 популяции лимфоцитов.
Дефицит субклассов IgG. ИДС развивается при дефиците каждого из подклассов, но при исследовании общего содержания IgG в крови редко об-наруживаются отклонения от нормальных значений, чаще оно в норме или повышено. Так как созревание клонов В-лимфоцитов, секретирующих IgG2 и IgG4, происходит не ранее 2-го года жизни, у детей раннего возраста имеется физиологический дефицит данных субклассов. Дефицит IgG2 обнаруживает-ся у 50% больных первичным ИДС, очень часто при общей вариабельной ги-поглобулинемии и, как правило, у детей старшего возраста проявляется хро-нической пневмонией и синдромом мальабсорбции. Селективный дефицит IgG1 может быть компенсирован за счет образования антител, относящихся к другим субклассам.
Изолированный дефицит IgA - одна из самых частых аномалий иммун-ной системы. Для него характерны низкое содержание IgA в сыворотке крови (менее 50 мг/л), отсутствие дефицита других классов иммуноглобулинов, нормальная способность организма к продукции антител, мало измененные показатели клеточного иммунитета. Так как IgA - основной иммуноглобулин системы местного иммунитета (секреторный IgA), обращают внимание на связь его дефицита с рецидивирующими и хроническими заболеваниями ды-хательных путей и ЛОР-органов. При отсутствии или низком содержании IgA в секретах создаются условия для развития аллергических и аутоиммун-ных заболеваний, предпосылки для развития дисбактериоза и воспалитель-ных заболеваний желудочно-кишечного тракта. С селективным дефицитом IgA может быть связано возникновение рецидивирующего герпетического стоматита, язвенного колита, регионального энтерита и др.
ИДС с поражением системы фагоцитоза
По механизму развития фагоцитарная недостаточность делится на три ос-новные формы:
Лейкопеническая - развивается вследствие подавления процессов проли-ферации и созревания моноцитов (ионизирующее излучение, ряд токсинов, цитостатики и др.) либо в результате наследственной блокады деления и дифференцировки, например миелоидной стволовой клетки.
Дисфункциональная - характеризуется расстройствами различных этапов процессов фагоцитоза и презентации антигена (подвижности фагоцитов, их адгезивных свойств, поглощения объекта фагоцитоза, переработки его и представления антигена лимфоцитам).
Дисрегуляторная - развивается вследствие нарушения регуляции различ-ных этапов фагоцитарной реакции биологически активными веществами (нейромедиаторами, гормонами, простагландинами, биогенными аминами, пептидами и др.).
Характеризуются нарушением дифференцировки стволовых клеток, бло-ком созревания Т- и В-лимфоцитов и их дефицитом. Комбинированные фор-мы иммунодефицита встречаются чаще, чем селективные. Как правило, они связаны с нарушением центральных органов иммунной системы. При комби-нированных ИДС ведущая роль принадлежит дефекту Т-клеток.
1. Cиндром ретикулярной дисгенезии - характеризуется уменьшением в костном мозге количества стволовых клеток. Характерна внутриутробная ги-бель плода, или дети гибнут вскоре после рождения.
Заболевание проявляется в первые месяцы жизни и часто характеризуется злокачественным течением. Наблюдается задержка прибавки массы тела, уже в первые дни жизни у некоторых детей появляются кореподобные высыпа-ния на коже, что может быть связано с реакциями несовместимости по отно-шению к материнским лимфоцитам, поступающим через плаценту в крово-ток ребенка. Развиваются признаки кожного кандидоза, диарея, острая ин-терстициальная пневмония, приобретающая затяжной и рецидивирующий характер. Дети очень восприимчивы к вирусным инфекциям. В крови выяв-ляется значительная лимфопения, особенно низко содержание Т-лимфоцитов. Содержание иммуноглобулинов всех классов заметно снижено. Исключение составляют грудные дети с IgG, полученными от матери. Патог-номоничны изменения вилочковой железы, гипоплазия миндалин и лимфа-тических узлов. Возникает неспособность проявлять реакции гиперчувстви-тельности замедленного типа. Дети редко доживают до 2-летнего возраста.
3. Синдром атаксии-телеангиэктазии (синдром Луи-Бар) обусловлен дефектом созревания, снижением функции Т-лимфоцитов, уменьшением их числа в крови (особенно Т-хелперов), дефицитом иммуно-глобулинов (осо-бенно IgA, IgE, реже IgG). Синдром характеризуется сочетанием атаксии и других неврологических отклонений с телеангиэктатическими изменениями сосудов склер, лица. Поражение нервной системы проявляется симптомами выпадения функций мозжечка, подкорковых ганглиев, диэнцефальной облас-ти, пирамидной системы. В результате их поражений возникают нарушение походки, замедленность произвольных движений, гиперкинезы, вегетососу-дистая дистония. У многих отмечаются вялотекущие пневмонии, развивают-ся ателектазы, пневмосклероз и бронхоэктазы. При исследовании лимфати-ческой системы устанавливается гипоплазия вилочковой железы, лимфатиче-ских узлов, селезенки. У многих детей отмечается уменьшение содержания в крови лимфоцитов, снижена реакция бласттрансформации лимфоцитов, не определяется IgA.
Заболевание характеризуется аутосомнорецессивным типом насле-дования.Прогноз синдрома неблагоприятен. Около 50% летальных исходов обусловлено хроническим поражением бронхолегочной системы, около 20% - развитием злокачественных процессов, которые связывают с утратой функ-циональной активности тимусзависимых лимфоцитов, а в общем плане – с отсутствием цензорной функции вилочковой железы – функции иммуноло-гического надзора. Некоторые больные доживают до 40-50 лет.
4. Синдром Вискотта-Олдрича характеризуется дефицитом перифери-ческих Т-лимфоцитов, нарушением их структуры и физико-химических свойств мембран, уменьшением клеточного иммунитета. Дефект, вероятно, связан с отсутствием на клеточной мембране лимфоцитов и тромбоцитов гликопротеида с относительной молекулярной массой 110 kD. При этом за-болевании выявляются снижение плотности микроворсинок на лимфоцитах, нарушение активации Т-лимфоцитов и не объясненная до сих пор нестабиль-ность экспрессии сиалогликопротеинов. Иммунологическая недостаточность обусловлена гипофункцией вилочковой железы. Часто обнаруживаются де-фицит IgM при нормальном содержании IgG и увеличение уровня IgA, IgE в крови. Характерно уменьшение продукции антител к антигенам-полисахаридам, но эти больные нормально реагируют на белковые антигены. Кроме того, имеют место врожденные дефекты тромбоцитов (нарушения ад-гезии, агрегации), тромбоцитопения. Болеют только мальчики. Заболевание проявляется в раннем возрасте, иногда в периоде новорожденности. Дети страдают частыми вирусными и бактериальными инфекциями. Характерны рецидивирующий гнойный отит, экзема, пиодермии, пневмония, колиты. Ге-моррагический синдром может быть ведущим.
Прогноз тяжелых форм неблагоприятен, дети погибают в возрасте до 10 лет. К летальному исходу приводят инфекции, геморрагии или злокачест-веннные новообразования лимфоретикулярной системы.
Недостаточность системы комплемента
Система комплемента представлена протеолитическими ферментами и ре-гуляторными белками. В крови имеются 20 комплементарных факторов, ак-тивация которых может осуществляться классическим или альтернативным путем. Активация комплемента обеспечивает защиту организма от любых чужеродных агентов; с активацией комплемента связаны и повреждающие эффекты при развитии аллергических и аутоиммунных реакций. При врож-денном и приобретенном расстройстве комплементарных факторов наруша-ются процессы фагоцитоза и происходит освобождение биологически актив-ных веществ.
При врожденном дефиците С1 невозможна активация системы компле-мента по классическому пути, поэтому, вследствие нарушения фаго-цитоза и лизиса микробов, наблюдаются повторные тяжелые гнойные процессы. При врожденном дефиците ингибитора С3б постоянно активируется комплемент С3, в результате чего содержание его в крови уменьшается. Хотя количество предшествующих комплементарных факторов (С1, С2, С4) не изменяется, однако из-за дефицита С3 нарушаются процессы фагоцитоза и лизиса бакте-рий, что проявляется повторными гнойными инфекциями.
При врожденном дефиците С5 склонность к инфекции также связана с на-рушением фагоцитоза и лизиса из-за невозможности образования соответст-вующих компонентов комплемента.
Содержание комплемента у детей на 30% ниже, чем у взрослых, что дела-ет понятным их склонность к инфекции и сепсису. Расстройства системы комплемента приобретенного характера проявляются в измене-нии количест-ва комплементарных факторов. При поражениях печени (цирроз, гепатит, хронический холецистит) нарушается синтез С1, С3, С6, С9. С другой сторо-ны, при аллергических, аутоиммунных заболеваниях комплементарные фак-торы уменьшаются в крови из-за связывания их иммунными комплексами.
Принципы лечения первичных ИДС
Лечение зависит от типа первичной иммунологической недостаточ-ности. Выделяются 3 основных направления иммунокоррекции.
Иммунная инженерия (трансплантация органов и тканей иммунной сис-темы: эмбриональной печени, тимуса, комплекса тимус-грудина, костного мозга, клеток иммунной системы; введение -глобулинов, иммуноглобулинов отдельных классов; сорбционные методы: гемосорбция, аффинная сорбция, иммуносорбция).
Коррекция гормонами и медиаторами иммунной системы (тимические гормональные факторы, миелопептиды, цитокины типа интерферона, интер-лейкины).
Фармакологическая коррекция - левамизол, диуцифон, полианионы и др.
Также применяется активная иммунизация против частых инфекций с по-мощью убитых вакцин, вводятся антибиотики, сульфаниламиды, противо-грибковые препараты.

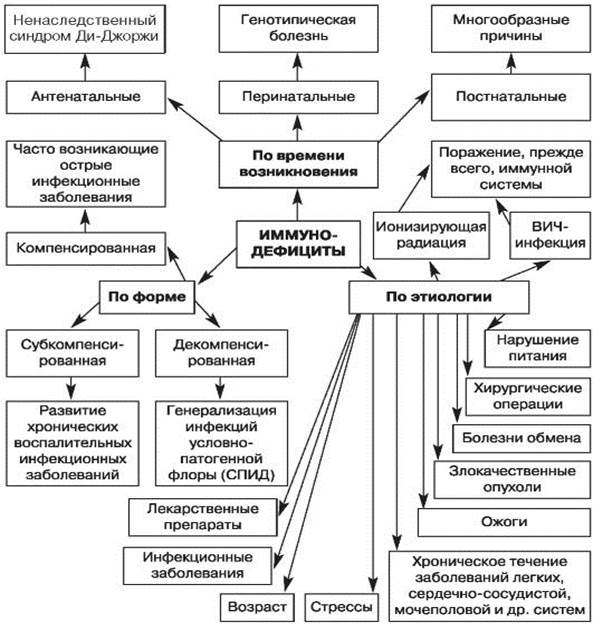
Рис. 1. Классификация вторичных иммунодефицитов по времени возникновения, этиологии, форме (по Белозерову Е.С. и др., 1992) [3]
Подводя итог в теоретической составляющей статьи – актуальности, необходимо обобщить следующее.
В педиатрии (17 лет 11 месяцев и 29 дней) в доминирующем большинстве речь идет об антенатальных и перинатальных иммунодефицитных болезнях, вторичных по отношению к анатомо-морфологическому субстрату иммунной системы – функциональных.
Таким образом, заключение врача-иммунолога должно быть сформулировано следующим образом. Антенатальная (или функциональная) иммунодефицитная болезнь, смешанного типа (или, при возможности, по отдельным звеньям, что мало вероятно), комбинированная индукция (инфекционная, гипоксическая и т.д.) с клиническими проявлениями основных синдромов – инфекционного, аллергического, аутоиммунного, неопластического и/или лимфопролиферативного.
Данные теоретические изыскания легли в основу целого цикла научно-исследовательских работ. Одно из таких исследований представлено в данной статье и посвящено исследованию иммуноиндуцирующей роли герпеса 4 типа - вируса Эпштейна-Барр у детей.
Цель: изучение особенностей клинических проявлений у детей с герпес-индуцированными формами иммунодефицитов (на примере 4 типа герпеса).
Все дети (100%) относились к часто и длительно болеющим, при этом 72 ребенка были неорганизованными (22,5%). У 78 (24,4%) детей были зарегистрированы признаки рецидивирующей герпетической инфекции с локализацией на губах, крыльях носа, слизистой оболочке полости рта. Ежегодно 92 ребенка (28,4%) переносили бактериальные инфекции в виде тонзиллитов, пневмоний, пиелонефритов, фурункулезов, гнойных конъюнктивитов, отитов, рино-синуситов. Признаки лимфоаденопатии (микролимфоаденопатии) регистрировались у 270 детей (84,4%). У 2 детей были диагностированы неопластические процессы (один ребенок умер в 2009 году). Одна девочка с лейкемоидной реакцией наблюдается у гематоонколога в течение 2 лет. Субфебрильные реакции более 12 месяцев были зарегистрированы у 39 детей (12,2%). Грубые неврологические нарушения (преимущественно двигательные) были зафиксированы у 43 детей (13,4%) (рис. 2).
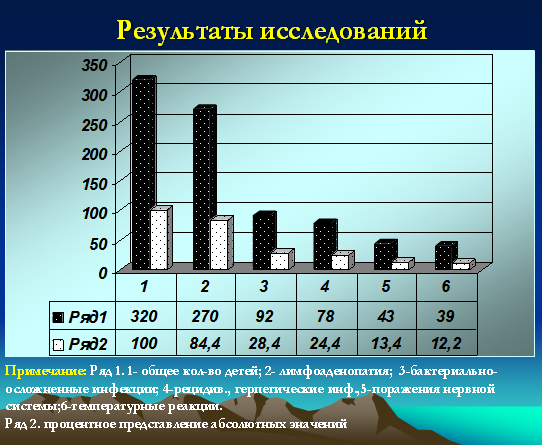
Рис. 2. Частота распределения клинических проявлений у детей с герпесиндуцированными иммунодефицитами
Иммунологические данные (в т.ч. данные лейкограммы), кроме количества лейкоцитов и лимфоцитов (абсолютное и относительное количество), носили неоднозначный характер. Классический супрессорный тип (в т.ч. и со снижением клеток цитолитической направленности) выявлялся у 108 детей (33,7%). Активация в системе клеток естественной цитотоксичности (CD16+) зафиксирована у 52 детей (16,2%). Увеличение В-клеточного пула было зарегистрировано у 38 детей (11,8%). Интересными, на наш взгляд, были полученные данные по CD95 маркеру, у 92 (28,7%) детей данный показатель был резко повышен и достигал значений 20-25%, у 30 (9,3%) детей указанный показатель не превышал 3% (рис. 3).
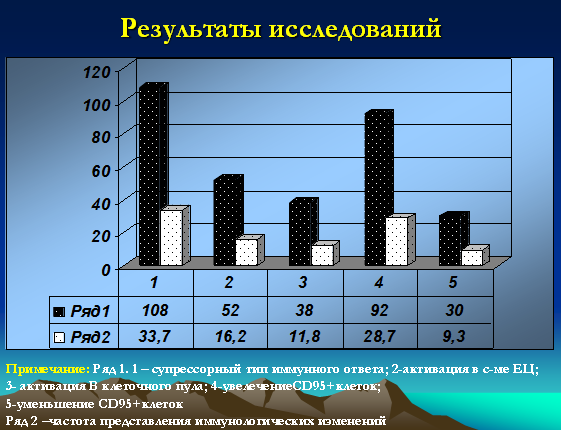
Рис. 3. Иммунологические особенности (типы распределения) у детей с герпесиндуцированными иммунодефицитами
ИФА, проведенный у детей, выявил 100% контаминацию вирусом ЭБ. Определялась следующая совокупность специфических антител:
- IgM к VCA (к капсидному антигену) – выявляются в крови в первые дни и недели болезни, максимально сохраняются до 3-4 недель;
- IgG к VCA (к капсидному антигену) – появляются в крови спустя 1-2 месяца от начала болезни, затем постепенно снижаются и сохраняются на пороговом (низком уровне) пожизненно. Повышение их титра характерно для обострения хронической ЭБВИ;
- IgM к EA (к раннему антигену) – появляются в крови в первую неделю заболевания, сохраняются в течение 2-3 месяцев и исчезают. Могут сохраняться в высоких титрах длительное время (более 3-4 месяцев), тревожно в плане формирования хронической формы ЭБВИ. Появление их при хронической инфекции служит индикатором реактивации;
- IgG к ЕA (к раннему антигену) – появляются к 3-4-й неделе заболевания, становятся максимальными на 4-6-й неделе болезни, исчезают через 3-6 месяцев. Появление высоких титров повторно указывает на активацию хронической инфекции;
- IgG к NA-1 или EBNA (к нуклеарному или ядерному антигену) – являются поздними, поскольку появляются в крови через 1-3 месяца после начала заболевания. При этом острофазные антитела выявлялись (IgM к VCA и IgM к EA) у 42 детей (13,1%), у 268 (83,7%) детей титры антител (IgG VCA + IgG к NA-1 или EBNA) значительно превышали нормативные и достигали значений более 160, у 10 (3,1%) детей диагностически значимых титров выявлено не было [9; 13] (рис. 4).
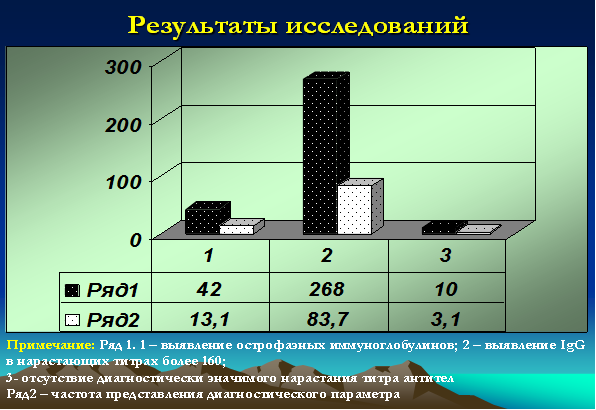
Рис. 4. Серологические исследования у детей с герпесиндуцированными иммунодефицитными болезнями
Клинические, иммунологические и этиологические сопоставления выявили следующие закономерности.
Отдельно хочется остановиться на методах ПЦР-диагностики. В литературе активно обсуждается вопрос об использовании ПЦР для исследований на инфекции семейства герпес в различных биологических субстратах. Здесь необходимо понимать, что данный метод, особенно при использовании его для исследования плазмы, имеет свои особенности – он всегда количественный! Определение качественное не имеет смысла, так как инфекция пожизненно персистирует в организме человека. Как у больных ЭБВИ, так и у носителей может быть положительная ПЦР. Поэтому для их дифференцировки проводится ПЦР-анализ с заданной чувствительностью: для носителей до 10 копий в пробе, а для активной инфекции – 100 копий в пробе. Специфичность данного метода хоть и достигает 100%, при этом не исключает ложноположительные результаты, т.к. ПЦР-анализ информативен только при размножении (репликации) вируса, то существует и определенный процент ложноотрицательных результатов с отсутствием репликации в момент исследования [5; 9].
В результате проведенного нами исследования, ПЦР крови всегда имела отрицательный результат, даже у детей с неопластическими процессами. Распределение ПЦР-положительных результатов у детей со слизистых оболочек (ротоглотка, нос) и мочи имело следующее цифровое распределение.
Положительные реакции с 3 биологических объектов имели 36 детей (11,25%), у 62 положительные ПЦР реакции были с зева и носа – 19,38%, у 43 детей диагностировали положительный результат только с зева (13.44%), и 71 ребенок имели положительные значения ПЦР со слизистой оболочки носа (22,19%). Положительные значения только в моче имели 51 ребенок (15,94%). Остальные дети (в количестве 57) имели отрицательный результат ПЦР-исследований с биологических сред.
Дети с неопластическими процессами, длительным субфебрилитетом, лейкемоидной реакцией, лимфоаденопатией имели признаки активации хронической инфекции, резкое увеличение CD95+ клеток, положительные маркеры наличия и обострения ВЭБ в ИФА и ПЦР-исследованиях.
Таким образом, все дети имели индуцированные формы функциональной иммунодефицитной болезни, доминирующей причиной которой являлся вирус ЭБ. Проведенные предварительные клинико-иммунологические исследования наглядно демонстрируют необходимость комплексного обследования детей с функциональными нарушениями в системе иммуногенеза с обязательной оценкой уровня инфекционной (вирусной) контаминации. Выявление основных причин трактуется необходимостью применения комплексных методов лечения, в т.ч. и с применением противовирусных препаратов.
Читайте также:
