
Признаки активации апоптоза на фоне вирусной инфекции что это
У РНК-содержащих ретровирусов сначала происходит обратная транскрипция генома в ДНК, затем ее интеграция в клеточные хромосомы и лишь после этого транскрипция генов.
Цитопатические эффекты при вирусных инфекциях разнообразны, они определяются как вирусом, так и клеткой и сводятся к разрушению клетки (цитолитический эффект), сосуществованию вируса и клетки без гибели последней (латентная и персистирующая инфекция) и трансформации клетки.
Вовлеченность организма в инфекционный процесс зависит от ряда обстоятельств - количества погибших клеток, токсичности вирусов и продуктов распада клеток, от реакций организма, начиная от рефлекторных и заканчивая иммунными. Количество погибших клеток влияет на тяжесть инфекционного процесса. Например, будут ли поражены при гриппе только клетки носа и трахеи или вирус поразит клетки эпителия альвеол, зависит тяжесть и исход болезни.
Хотя вирусы и не образуют типичных токсинов, однако и вирионы, и вирусные компоненты, накапливающиеся в пораженных тканях, выходя в кровоток, оказывают токсическое действие. Неменьшее токсическое действие оказывают и продукты распада клеток. В этом случае действие вирусной инфекции столь же неспецифично, как и действие патогенных организмов, убивающих клетки и вызывающих их аутолиз. Поступление токсинов в кровь вызывает ответную реакцию - лихорадку, воспаление, иммунный ответ. Лихорадка является преимущественно рефлекторным ответом на поступление в кровь и воздействие на ЦНС токсичных веществ.
Если лихорадка - общий ответ организма на вирусную инфекцию, то воспаление - это местная многокомпонентная реакция. При воспалении происходят инфильтрация пораженных тканей макрофагами, утилизация продуктов распада, репарация и регенерация. Одновременно развиваются реакции клеточного и гуморального иммунитета. На ранних стадиях инфекции действуют неспецифические киллеры и антитела класса IgM. Затем вступают в действие основные факторы гуморального и клеточного иммунитета. Однако гораздо раньше, уже в первые часы после заражения, начинает действовать система интерферона, представляющая семейство секреторных белков, вырабатываемых клетками организма в ответ на вирусы и другие стимулы. Описанные явления относятся к так называемой острой репродуктивной вирусной инфекции. Взаимодействие вируса и клеток может происходить, как отмечалось выше, без гибели последних. В этом случае говорят о латентной, т.е. бессимптомной или персистирующей хронической вирусной инфекции. Дальнейшая экспрессия вируса, образование вирусспецифических белков и вирионов вызывает синтез антител, на этой стадии латентная инфекция переходит в персистирующую и появляются первые признаки болезни.
Репродукция вируса в клетках сопровождается развитием цитопатических процессов, специфичных для разных вирусов и для разных типов инфекционных процессов. Цитопатические процессы при вирусных инфекциях разнообразны, они определяются как вирусом, так и клетками, причем специфика их больше "задается" клеткой, нежели вирусом, и сводится в основном к разрушению клеток, сосуществованию вируса и клеток без гибели последних и трансформация клеток. Несмотря на значительные различия цитоцидного действия разных вирусов, в общем, они сходны. Подавление синтеза клеточных макромолекул - нуклеиновых кислот и белков, а также истощение энергетических ресурсов клетки ведут к необратимым процессам, заканчивающимся гибелью пораженной клетки. Повреждение клеток вирусами, их отмирание и распад переносят вирусную инфекцию с клеточного уровня на уровень организма в целом.
При встрече организма с вирусной инфекцией продукция интерферона (растворимого фактора, вырабатываемого вирус-инфицированными клетками, способного индуцировать антивирусный статус в неинфицированных клетках) становится наиболее быстрой реакцией на заражение, формируя защитный барьер на пути вирусов намного раньше специфических защитных реакций иммунитета, стимулируя клеточную резистентность, - делая клетки непригодными для размножения вирусов.
Продукция и секреция цитокинов относятся к самым ранним событиям, сопутствующим взаимодействию микроорганизмов с макрофагами. Этот ранний неспецифический ответ на инфекцию важен по нескольким причинам: он развивается очень быстро, поскольку не связан с необходимостью накопления клона клеток, отвечающих на конкретный антиген; ранний цитокиновый ответ влияет на последующий специфический иммунный ответ.
Интерферон активирует макрофаги, которые затем синтезируют интерферон-гамма, ИЛ-1, 2, 4, 6, ФНО, в результате макрофаги приобретают способность лизировать вирус-инфицированные клетки.
Интерферон-гамма является специализированным индуктором активации макрофагов, который способен индуцировать экспрессию более 100 разных генов в геноме макрофага.
Продуцентами этой молекулы являются активированные Т-лимфоциты (Тh1-тип) и естественные киллеры (NK-клетки). Интерферон-гамма индуцирует и стимулирует продукцию провоспалительных цитокинов (ФНО, ИЛ-1, 6), экспрессию на мембранах макрофагов, антигенов МНС II; гамма-интерферон резко усиливает антимикробную и противовоспалительную активность путем повышения продукции клетками супероксидных радикалов, а усиление иммунного фагоцитоза и антителоопосредованной цитотоксичности макрофагов под влиянием гамма-интерферона связано с усилением экспрессии Fc-рецепторов для JgG. Активирующее действие интерферона-гамма на макрофаги опосредовано индукцией секреции этими клетками ФНО -альфа. Этот пик наблюдается совместно с ФНО-альфа. Максимум продукции ИЛ-4 наступает через 24-48 ч с момента активации клеток. При этом ИЛ-4 рассматривается как цитокин, ограничивающий иммуновоспалительные реакции и снижающий ответ организма на инфекцию, угнетая при этом экспрессию гамма-интерферона. Интерферон-гамма ин витро усиливает фагоцитарную активность нейтрофилов, что обусловлено усилением экспрессии Fc-рецепторов и поверхностных белков семейства интегринов на нейтрофилы. Это позволяет нейтрофилам осуществлять цитотоксические функции и фагоцитоз. В качестве основных эффекторных клеток воспалительного процесса, они обеспечивают элиминацию инфекта из организма.
Взаимодействие цитокина с клеткой определяется универсальной биологической системой, специфическим механизмом которой является рецепторный аппарат, связанный с восприятием метаболического кода. Для проявления биологической активности цитокина необходимо присутствие на поверхности чувствительных клеток специфических рецепторов, которые могут экспрессироваться параллельно с синтезом цитокина. Рецепторы цитокинов представляют собой комплексы, состоящие из двух и более рецепторных молекул, которые объединяются на мембране клетки-мишени и образуют высокоаффинный рецепторный комплекс. Большинство рецепторов состоит из отдельных молекул, связывающих цитокины, которые ассоциируются после связывания лиганда с сигналпередающим рецепторным компонентом; часть рецепторов существует как растворимые изоформы, способные связывать и растворять цитокины, а часть функционирует как многокомпонентные блоки; механизм комплексирования субъединиц рецепторов объясняет плейотропные и дублирующие эффекты цитокинов, имеющих большое структурное сходство. Рецепторы ИЛ-10 имеют гомологию рецепторов интерферона, и подобно ИЛ-10 индуцирует экспрессию в моноцитах гена Fc- рецептора. Для полного функционирования цитокиновой системы необходимы повышение уровня цитокина в ответ на инфект и экспрессия нормального количества рецепторов к ним на клетках. Изменение рецепторов после их связывания с цитокином заключается в интернализации комплексов цитокин - рецептор внутрь клетки. На поверхности клеток рецептор появляется заново, постепенно синтезируясь в течение 24-36 ч (время появления рецепторов интерферон-альфа). В этот период клетки остаются чувствительными к последующим дозам цитокина, чем объясняется эффективность введения препаратов интерферона и их индукторов три раза в неделю.
Пик продукции цитокинов после стимуляции макрофагов наблюдается через 1-2,6,18-48 ч, а пик продукции интерферон-гамма наступает через 20 ч после первого выхода цитокина из клетки. Поскольку интерферон-гамма ингибирует миелопоэз, то нормализация числа нейтрофилов после элиминации инфекта связана с системой регуляции нейтропоэза. Через 6 ч после стимуляции интерферон-альфа для выполнения своих функций NK-rклетки (активность которых регулируется ИЛ-1, 4, 2) продуцируют гамма-интерферон, в результате чего происходит лизис инфицированных клеток.
При антигенной стимуляции клеток трансдукция сигнала с активированного рецептора на генетический аппарат осуществляется с помощью внутриклеточных регуляторных систем, компоненты которых (белки мембран, ферментов, хроматина) связываются с чувствительными к ним последовательностями ДНК. После связывания цитокина (интерферон) с поверхностными клеточными мембранными рецепторами происходит активация ферментов протеинкиназы-С (ПКС), тирозинкиназы, ц-АМФзависимой протеинкиназы, серин-треонинкиназы. Интерферон-альфа активирует tyk 2 и jak 1-киназы, а интерферон-гамма активирует jak 1 и 2-киназы. Далее факторы транскрипции перемещаются в ядро клетки и связывают гены раннего ответа.
Первый ответ клеток на цитокин - это быстрая индукция генов раннего ответа ("immediate early" генов), в число которых и входит ген интерферон-гамма. Стимуляция экспрессии этих генов важна для выхода клеток из Go-стадии и перехода в Gi-стадию и дальнейшей прогрессии клеточного цикла. Их индукция происходит после активации рецепторов роста на клеточной мембране и активации протеин-киназной системы. Гены раннего ответа являются ключевыми регуляторами клеточной пролиферации и дифференцировки, кодируют белки, регулирующие репликацию ДНК.
Таким образом, при активации клеток происходит стимуляция генов раннего ответа, что ассоциируется с изменением фаз клеточного цикла. Основная протективная роль в иммунном ответе, направленном против внутриклеточных паразитов (грибы, простейшие, вирусы, микобактерии туберкулеза), принадлежит клеточным механизмам. Способность перечисленных возбудителей переживать и размножаться внутри клеток делает их защищенными от действия антител и системы комплемента. Резистентность к антимикробным факторам макрофагов позволяет им длительно переживать внутри этих клеток. Для элиминации возбудителя необходим специфический клеточно-опосредованный ответ. Его специфичность определяется антигенраспознающими СД8+-Т-лимфоцитами, которые пролиферируют, активируются и формируют клон эффекторных цитотоксических лимфоцитов. Решающий момент специфического иммунного ответа - это ответ СД4+Т-лимфоцитов с хелперной направленностью на распознавание антигена. На этом этапе определяется форма иммунного ответа: либо с преобладанием гуморального иммунитета, либо с преобладанием клеточных реакций (ГЗТ). Направление дифференцировки СД4 + -лимфоцитов, от которого зависит форма специфического иммунного ответа, контролируется цитокинами, образующимися в ходе воспалительной реакции. Так, в присутствии ИЛ-12 и интерферон-гамма СД4 + -лимфоциты дифференцируются в воспалительные Тh1-клетки, начинают продуцировать и секретировать интерлейкин-2, интерферон-гамма, ФНО и определяют клеточный характер специфического иммунного ответа. Присутствие ИЛ-12 обеспечивается его продукцией макрофагами, а интерферон-гамма - естественными киллерами, активированными в раннюю фазу ответа на внутриклеточно паразитирующие бактерии и вирусы. В отличие от этого, в присутствии ИЛ-4 СД4 + -лимфоциты дифференцируются в хелперы Тh 2, которые начинают продуцировать и секретировать ИЛ-4, ИЛ-5, ИЛ-6 и запускают гуморальный иммунный ответ, т.е. синтез специфических антител - иммуноглобулинов. Воспалительные Тh 1-лимфоциты нужны для борьбы с внутриклеточными паразитами, а Тh 2 хелперы нужны для элективной защиты от внеклеточных паразитов.
Вирусная инфекция может вызывать быстрое подавление экспрессии ряда клеточных генов (из которых наиболее изучены интерфероновые гены и гены, кодирующие дс-РНК-зависимые ферменты -2,5-ОАС и ПК-дс), принимающих участие в антивирусном действии. Специальные исследования механизма антивирусного действия интерферонов и дс-РНК в клеточных и бесклеточных системах показали ключевую роль в этом процессе вышеуказанных ферментов. ПК-дс, взаимодействуя с дс-РНК, фосфорилируется и в активной форме фосфорилирует регуляторные факторы транскрипции и трансляции, из которых наиболее изучен инициирующий фактор трансляции (eIF2).
ПК-дс выполняет регуляторную роль в системе клеточной пролиферации на уровне факторов трансляции и активации ряда генов цитокинов. Вероятно, существует связь между подавлением транскрипции мРНК и ПК-дс, угнетением общего синтеза клеточного белка при вирусных инфекциях и накоплением в ядрах клеток белка нуклеокапсида и белка NSP2. Фрагментация клеточных хромосом, наблюдающаяся на ранних сроках вирусной инфекции, может быть одной из причин подавления экспрессии генов, участвующих в противовирусном ответе.
Есть основания предполагать участие белков NSP2 в регуляции активности генов цитокинов - низкомолекулярных белковых регуляторных веществ, продуцируемых клетками и способных модулировать их функциональную активность. Нарушения в системе цитокинов приводят к нарушению кооперативных взаимодействий иммунокомпетентных клеток и нарушению иммунного гомеостаза.
В последние годы показано, что ИЛ- 12, относящийся к провоспалительным цитокинам, является ключевым для усиления клеточно-опосредованного иммунного ответа и инициации эффективной защиты против вирусов.
Средства терапии гриппа и ОРЗ можно разделить на этиотропные, иммунокорригирующие, патогенетические и симптоматические. Приоритет принадлежит этиотропным препаратам, действие которых направлено непосредственно на возбудитель инфекции. Все препараты этиотропного действия целесообразно рассматривать с учетом их точек приложения в цикле репродукции вирусов гриппа и других ОРЗ.
Применение химиопрепаратов для профилактики и лечения гриппа и ОРЗ относится к базовой терапии и является общепризнанным мировым стандартом. Многолетние клинические исследования достоверно выявили их высокую лечебно-профилактическую значимость. Химиотерапевтические средства представлены тремя основными группами: это блокаторы М2-каналов (амантадин, ремантадин); ингибиторы нейраминидазы (занамивир, озельтамивир) и ингибиторы протеаз (амбен, аминокапроновая кислота, трасилол). Препараты оказывают прямое антивирусное действие, нарушая различные фазы репликативного цикла вирусов. Несколько особняком стоит группа вирулицидных препаратов, применяемых местно для предотвращения адсорбции и проникновения вирионов в клетки.
- Грипп и другие респираторные вирусные инфекции / под ред. О.И. Киселева, И.Г. Мариничева, А.А. Сомининой. - СПб, 2003.
- Дриневский В.П., Осидак Л.В., Цыбалова Л.М. Острые респираторные инфекции у детей и подростков // Практическое руководство под редакцией О.И. Киселева. - СПб, 2003.
- Железникова Г.Ф., Иванова В.В., Монахова Н.Е. Варианты иммунопатогенеза острых инфекций у детей. СПб, 2007. - 254 с.
- Ершов Ф.И. Грипп и другие ОРВИ // Антивирусные препараты. Справочник. - М., 2006. - С.226-247.
- Ершов Ф.И., Романцов М.Г. Антивирусные средства в педиатрии. - М., 2005. - С.159-175.
- Ершов Ф.И., Киселев О.И. Интерфероны и их индукторы (от молекул до лекарств). М., 2005. - С.287-292.
- Иванова В.В. Острые респираторно-вирусные заболевания // Инфекционные болезни у детей. - М., 2002.
- Онищенко Г.Г., Киселев О.И., Соминина А.А. Усиление надзора и контроля за гриппом как важнейший элемент подготовки к сезонным эпидемиям и очередной пандемии. - М., 2004. - С.5-9.
- Об утверждении стандарта медицинской помощи больным гриппом, вызванным идентифицированным вирусом гриппа (грипп птиц) // Приказ Минздравсоцразвития №460 от 07.06.2006 г.
- Романцов М.Г., Ершов Ф.И.Часто болеющие дети: Современная фармакотерапия. - М., 2006. - 192 с.
- Стандартизированные принципы диагностики, лечения и экстренной профилактики гриппа и других острых респираторных инфекций у детей / под ред. О.И. Киселева. - Санкт-Петербург. - 2004. - С.82-95.
- Лекарственные средства в фармакотерапии патологии клетки / под ред. Т.Г.Кожока. - М., 2007.
Несмотря на высокий уровень развития цивилизации, бремя травматизма остается неразрешенной проблемой, которая продолжает привлекать пристальное внимание клиницистов и исследователей, приводит к большим потерям даже в экономически благополучных странах, и входит в первую тройку ведущих причин смерти [1]. Частота алкогольного опьянения у лиц, пострадавших в дорожно-транспортных происшествиях (при смертельном и несмертельном травматизме), достигает 30–40 %. Алкогольное опьянение, в котором нередко находятся пациенты, значительно утяжеляет их состояние, затрудняет диагностику и оказание неотложной помощи на догоспитальном этапе. Алкогольная интоксикация изменяет клиническую картину травмы. Изучение патогенетических особенностей при сочетанной травме в сочетании с алкоголизацией показало, что больные в состоянии алкогольного опьянения должны рассматриваться как группа повышенного риска, поскольку их компенсаторные механизмы ослаблены [9,10]. Терапевтическая тактика в отношении травмированных пациентов с алкогольными проблемами имеет свои особенности. Однако не всегда можно получить достоверную информацию о наличии алкогольных проблем у пациента. При тяжелых сочетанных травмах зачастую невозможно определить донозологическое состояние пострадавшего, и ориентироваться приходится на косвенные признаки. Поиск информативных методов диагностики остается актуальной задачей.
Алкогольная интоксикация, предшествующая травме, вызывает метаболические нарушения, токсическое поражение печени, которые утяжеляют прогноз. Одним из механизмов неудовлетворительных репаративных процессов при хронической алкогольной интоксикации может явиться усиление процессов апоптоза.
В настоящее время используются два определения апоптоза: морфологическое, в котором подразумевается апоптоз как форма клеточной гибели, проявляющаяся в её уменьшении, в фрагментации и конденсации хроматина и в уплотнении цитоплазматической мембраны, без выхода содержимого в межклеточное пространство; и биохимическое определение, при котором апоптоз – это программируемая клеточная смерть в результате реализации генетической программы или ответа на внешние факторы, требующая энергетических затрат и синтеза новых макромолекул [6]. В настоящее время под апоптозом принято понимать каспазозависимый процесс упорядоченной гибели отдельных клеток, который происходит в нормальных и патологически измененных органах и тканях организма под действием внеклеточных или внутриклеточных стимулов [8].
В современной классификации программированной смерти клеток (ПСК) выделяют ПСК I типа – апоптоз, II типа – аутофагию и III типа – некроз клетки (онкоз) [6]. Принципиальное различие между апоптозом и некрозом заключается в различных последствиях этих процессов для окружающих тканей и всего организма. Апоптоз завершается фагоцитозом апоптотических телец макрофагами или стромальными клетками, не происходит явлений альтерации, воспаления или иммунного ответа, тогда как при некрозе наблюдаются все эти процессы, развивается полноценный иммунный ответ [2].
Программа апоптотической гибели клетки проходит несколько этапов:
- активация про-апоптотических белков,
- активация каскада белковых комплексов – каспаз, расщепляющих белки-мишени,
- морфологическая перестройка внутриклеточных органелл.
- фрагментация клетки с образованием апоптотических телец,
- фагоцитоз макрофагами или стромальными клетками [5,6].
При каспазном механизме клеточной гибели происходит следующее. Активируются инициаторные, затем – эффекторные каспазы. Последние и активированные ими ферменты (эндонуклеазы) разрушают компоненты клетки, фрагментируют ДНК, в результате хроматин скапливается по периферии ядра, распадается цитоскелет, митохондрии, аппарат Гольджи, эндоплазматический ретикулум. При некаспазном механизме клеточной гибели из митохондрий выходят флавопротеин AIF и эндонуклеаза G, мигрируют в ядро и вызывают распад ядерной ДНК на олигонуклеотиды. Наблюдается конденсация хроматина и экспозиция фосфатидилсерина во внешнем монослое плазматической мембраны. Конечный этап – фрагментация ядра, клетки и образование апоптотических телец [6,7].
Стандартного метода выявления апоптоза не существует. Используются следующие методы морфологической верификации в комплексе:
- рутинная микроскопия с использованием обычных методов фиксации и окрашивания;
- выявление олигонуклеосомной деградации ДНК в отдельных клетках.
Метод электронной микроскопии является наиболее точным, однако из-за необходимости идентификации процесса апоптоза в отдельных клетках он является слишком затратным для массового применения. Кроме того, по оценкам, отдельные стадии программируемой клеточной гибели длятся не более 1–1,5 часов, что также затрудняет их выявление.
Формально наиболее вероятным критерием апоптоза является наличие каспазозависимой олигонуклеосомной деградации хроматина при сохранении целостности мембранных структур клеток [2,3], но практически это можно выявить лишь в специально оснащённых лабораториях.
Апоптоз не является специфической реакцией на какие-то определённые патологические процессы. Так, согласно исследованиям E.Mutijima и др. (2014), при морфологическом исследовании операционного материала шейки бедра TUNEL-методом апоптотические остеоциты и остеобласты выявляется одинаково часто как при остеоартритах, так и при переломах, алкоголь-индуцированных или идиопатических остеонекрозах, в целом, независимо от этиологического фактора [23].
Хотя апоптоз и является нормальным саногенетическим процессом, в некоторых случаях тяжелого повреждения он носит характер патологического и усугубляет состояние пострадавшего [15,16,17].
При черепно-мозговой травме (ЧМТ) повреждение ткани головного мозга и экстрацеребральные нарушения вызывают метаболические сдвиги и запускают патологические процессы, приводящие к гибели нервных клеток через механизм некроза и апоптоза. При некрозе происходит разрушение мембраны и внутриклеточных структур лизосомальными ферментами и массовая гибель клеток. Активируется система цитокинов, происходит повреждение соседних клеток, включается иммунный ответ. Повреждённые очаги хорошо определяются с помощью рутинных способов визуализации. Проявления апоптоза не столь очевидны и более отсрочены по времени от момента травмы, чем некроз. Для активации механизма апоптоза при ЧМТ необходимо образование ложных нейромедиаторов и нейротоксичных аминокислот. Предположительно считается, что степень активации нейротоксического каскада пропорциональна тяжести повреждения мозга, а непосредственным повреждающим фактором является гипоксия [12]. Активация каспаз при апоптозе приводит к постепенной гибели нейрона, сморщиванию клетки и выпадению её функции. Поскольку гибнут отдельные клетки, а соседние с ними нейроны не страдают, и при этом не развивается системной реакции воспаления, определить объёмы апоптотически погибших нервных клеток клиническими методами визуализации невозможно. Вместе с тем, установлены общие антигенные детерминанты белков мозга и тимуса человека [7,8], т.е., если по картине периферичекой крови можно судить о состоянии тимуса и иммунной системы, то по этим же показателям можно получить представление о состоянии мозговой ткани.
Не только гипоксия тканей головного мозга действует повреждающе, но и избыток кислорода, что вызывает активацию перекисного окисления и повреждение белков и липидов клеточной мембраны. Для нейтрализации этих процессов предложено использовать препараты с антиоксидантной активностью, например, производные янтарной кислоты, которые, кстати, широко применяются в комплексной терапии алкоголизма. Однако их клиническая эффективность в настоящее время не является строго доказанной [12]. Некоторые авторы рассматривают этанол в качестве нейропротективного агента при инсульте или ЧМТ. Так, в недавнем исследовании американских учёных, проведённом на крысах, было показано, что введение этанола в небольшой дозе после инсульта предупреждает развитие обширных повреждений головного мозга. Введение этанола в дозе 1,5 г/кг массы тела животного в срок до 4 часов после двухчасовой окклюзии средней мозговой артерии оказывает сильное нейропротективное действие, повышает экспрессию гипоксия-индуцированного фактора 1α и предупреждает внутримозговое кровоизлияние после использования тромболитиков [14, 24]. В другом исследовании, проведённом в Турции, изучено влияние умеренных доз алкоголя на цистеин-протеаза-индуцированный апоптоз нейронов и образование оксида азота в тканях мозга при повреждении. В группе крыс, получивших этанол и травму мозга, уровень активности ферментов каспазы-3 и катепсина-L оказался ниже, чем у крыс, получивших только травму. Авторы объясняют это способностью этанола подавлять активность лизосомальных протеаз и продукцию оксида азота [17].
Однако использование этанола в качестве нейропротектора представляется чрезвычайно спорным шагом. Хроническое употребление алкоголя увеличивает риск развития острого респираторного дистресс-синдрома, тяжелой формы острого повреждения легких, которое характеризуется поражением альвеолярного эпителия и эндотелиального барьера и развитием интенсивного воспаления. Злоупотребление алкоголем ассоциировано с большей частотой развития сепсиса или пневмонии, увеличением времени пребывания в стационаре, медицинских расходов и более высокой смертностью [9,18].
Нейродегенеративное действие алкоголя доказано во множестве исследований. Этанол-индуцированные повреждения лежат в основе многих расстройств алкогольного спектра. По мнению Luo J. (2012), этанол не подавляет, а индуцирует апоптоз [20]. Схематически это показано на рисунке.
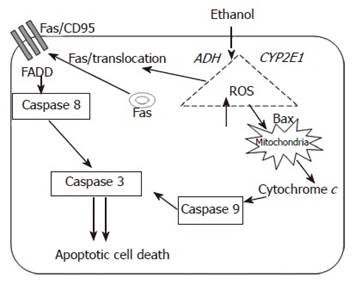
Схема инициации и распространения этанол-индуцированного апоптоза
Метаболизм этанола и сопутствующая генерация активных форм кислорода активируют про-апоптотические механизмы через рецептор смерти Fas/CD95 и внутренние механизмы апоптоза [22].
Другие исследования показали, что употребление алкоголя перед ожоговой травмой ослабляет иммунную защиту и повышает вероятность развития инфекционных осложнений. В тканях селезёнки у получивших алкоголь мышей уменьшалось образование интерлейкина-2 и снижалась продукция Т-лимфоцитов, по сравнению с также травмированными мышами, не получавшими алкоголь [15].
Основными физиологическими регуляторами клеточной гибели являются глюкокортикоиды. Дегидроэпиандростерон (ДГЭА) и его конъюгированная форма ДГЭА-сульфат (ДГЭАС) является ключевым звеном в биосинтезе всех стероидных гормонов и обладает собственными эффектами. Этот нейростероид оказывает нейропротективное и стресспротективное действие, защищая организм от пагубного воздействия высоких доз кортизола [11]. Нейростероиды обладают многими эффектами в ЦНС и могут быть вовлечены в патофизиологические процессы при травме, а также при алкоголизме. Оценка соотношения концентраций ДГЭА и кортизола характеризует состояние анаболических и катаболических процессов, т.е. стресслимитирующих и стрессреализующих систем, что можно использовать в прогнозе реабилитационного процесса.
Применение кортикостероидов при ЧМТ для лечения и предупреждения отека мозга теоретически должно приводить к стабилизации мембраны нервной клетки, предупреждению отёка и гибели клетки, а также к подавлению избыточной активации воспалительных цитокинов, развивающейся вследствие некроза тканей. Большие дозы кортикостероидов эффективно уменьшают сосудистый отек вокруг очагов повреждения. В крупном проспективном исследовании сверхраннего применения кортикостероидов при ЧМТ (CRASH trial colloborators, 2004, n>10000) показано, что отрицательные эффекты кортикостероидов, в случаях, когда они, возможно, были показаны, превышали предполагаемую пользу. Результаты лечения были хуже, а летальность – выше в группах, получавших метилпреднизолон. В исследование не включались те случаи, когда кортикостероиды были определённо показаны или определённо не показаны. В клинической практике их можно использовать для экстренного повышения церебрального перфузионного давления вместе с симпатомиметиками и инфузионной терапией [12].
Несмотря на то, что считается доказанным участие апоптоза в развитии отсроченной гибели нейронов головного мозга после травматического повреждения, вопрос о биологической роли этого процесса остается открытым. Пока до конца не известно, является ли программируемая гибель клетки исключительно вредным процессом или же устранение нефункционирующих клеток, находящихся в состоянии парабиоза, является приспособительной реакцией, направленной на усиление выживаемости, когда в условиях ЧМТ повышено внутричерепное давление, нарушена микро- и макроциркуляция, увеличена проницаемость гематоэнцефалического барьера. Безусловно, продолжение исследований данной проблемы обосновано и актуально.
Что касается апоптотических процессов в нейтрофилах человека с тяжелой скелетной и черепно-мозговой травмой, то на основании их учета можно сделать заключение, как минимум, о стадии иммунного дистресс-синдрома при критических состояниях [5].
Целью данного исследования было изучение признаков апоптоза нейтрофилов у пострадавших с сочетанными черепно-мозговыми и скелетными травмами (СЧМСТ) и гормональной регуляции, для возможного использования этих показателей в прогнозе состояния потерпевшего.
Материал и методы исследования. Обследованы пострадавшие с СЧМСТ, всего 58 чел., из них 30 чел. – в состоянии алкогольной интоксикации и с признаками хронического алкоголизма (1 группа), и 28 чел. – без признаков алкоголизма и алкогольной интоксикации (2 группа). Все пациенты были мужского пола, возраст от 31 до 57 лет, в среднем 41,52±1,25 года. У всех пострадавших состояние было квалифицировано как средней степени тяжести. Забор крови проводился при поступлении пациентов в клинику, до оперативного или консервативного лечения и до назначения фармакотерапии. В качестве контрольной группы использовались образцы крови 22 соматически и психически здоровых лиц, соответствующие по полу и возрасту обследуемой группе пациентов, не имеющих хронических заболеваний и не состоящих на диспансерном учете, без признаков перенесенных острых инфекционных заболеваний на момент обследования.
Для оценки апоптоза использовали световую микроскопию, поскольку результаты морфологического анализа хорошо коррелируют с данными других методов оценки апоптоза [3]. Посредством световой микроскопии окрашенных мазков из кондиционированной венозной крови определяли уровень апоптотических и некротически измененных нейтрофилов.
Результаты и их обсуждение
Для лимфоцитов с признаками апоптоза характерны деградация ядерного материала и фрагментация хроматина на несколько частей. Уровень спонтанного апоптоза лимфоцитов в мазках, приготовленных сразу после взятия крови, у травмированных лиц с признаками алкоголизма (1 гр.) достоверно отличался от значений, наблюдаемых у травмированных без признаков алкоголизма (2 гр.).
Цитологический анализ мазков крови показал, что у лиц 1 группы уровень спонтанного апоптоза лимфоцитов составляет 5,6±0,7 % (в контрольной группе здоровых лиц 0,9±0,2 %, у лиц 2 группы 2,4±0,4 %, p 0,05). У лиц 1 группы выявлено повышение экспрессии рецептора CD95 (16,9±0,3 %, во 2 гр. – 11,6±0,3 %, p
Читайте также:
