
Лейкоэнцефалопатия с neuroaxonal с сфероидам
, MD, University of Utah School of Medicine
Last full review/revision January 2019 by John E. Greenlee, MD

Этиология
Прогрессируящая мультифокальная лейкоэнцефалопатия развивается вследствие реактивации повсеместно распространенного JC-вируса из семейства паповавирусов, который обычно попадает в организм в детстве и пребывает в латентном состоянии в почках и других органах и тканях (например, в мононуклеарных клетках, ЦНС). Реактивированный вирус тропен к олигодендроцитам.
Большинство пациентов с развивающейся ПМЛ, имеют ослабление клеточного иммунитета, вызванное:
СПИД (наиболее распространенный фактор риска)
Расстройства ретикулоэндотелиальной системы (например,, лейкемия, лимфома)
Риск развития ПМЛ у больных СПИДом возрастает с увеличением вирусной нагрузки; в настоящее время распространенность ПМЛ уменьшилась в связи с широким применением более эффективных антиретровирусных препаратов.
В то же время возрастает количество случаев ПМЛ, таких как осложнения иммуномодулирующей терапии (например, моноклональными антителами, такими как натализумаб и ритуксимаб). Измерение сывороточных антител к вирусу JC (индекс вируса JC) может помочь оценить риск ПМЛ у пациентов, принимающих натализумаб.
Клинические проявления
Первым проявлением заболевания может быть неловкость и неповоротливость при движениях. Очень часто при обследовании выявляется гемипарез. Также часто наблюдаются афазия, дизартрия и гемианопсия. У двух третей больных многоочаговое поражение коры больших полушарий приводит к развитию когнитивных нарушений. Могут наблюдаться поражения сенсорных структур, мозжечка и ствола головного мозга.
Головные боли и судорожные припадки редки и встречаются чаще всего у больных СПИДом.
Неуклонное прогрессирование заболевания приводит к смерти, как правило, спустя 1–9 мес. после появления симптомов заболевания.
Диагностика
Исследование ЦСЖ на ДНК JC-вируса
Наличие прогрессирующей мультифокальной лейкоэнцефалопатии следует подозревать у пациентов с необъяснимыми прогрессирующими признаками поражения головного мозга, особенно у больных с иммунодефицитными состояниями.
Предварительный диагноз ПМЛ ставится на основании данных МРТ с контрастом, выявившей единичные или множественные очаги поражения белого вещества на Т2-взвешенных изображениях. Контрастное вещество слабо накапливается, как правило, по периферии в 5–15% очагов. При КТ могут определяться очаги пониженной плотности, не накапливающие контраст, однако этот метод является менее чувствительным по сравнению с МРТ.
ЦСЖ исследуется на ДНК JC-вируса с использованием ПЦР; положительные результаты этого анализа в сочетании с характерными изменениями при нейровизуализации подтверждают диагноз ПМЛ. Общий анализ ЦСЖ, как правило, не изменен.
Серологические исследования неинформативны. Иногда с целью дифференциальной диагностики выполняют стереотактическую биопсию мозга, которая, впрочем, редко себя оправдывает.
Лечение
Лечение прогрессивной многоочаговой лейкодистрофи, в основном, поддерживающее.
Экспериментальное применение цидофовира и других противовирусных препаратов не подтвердило своей эффективности. Антиретровирусная терапия (АРТ) улучшает прогноз заболевания у ВИЧ-инфицированных пациентов с ПМЛ, увеличивая уровень годовой выживаемости с 10 до 50%. В то же время, агрессивная антиретровирусная терапия может приводить к развитию у пациентов воспалительного синдрома восстановления иммунной системы (ВСВИС). При ВСВИС восстанавление иммунной системы вызывает интенсивную воспалительную реакцию на вирус JC, тем самым ухудшая симптоматику. При проведении нейровизуализации на фоне ВСВИС выявляются значительное усиление накопления контрастного вещества очагами и иногда – отек головного мозга. Эффективной может быть терапия кортикостероидами. В зависимости от тяжести течения ВСВИС и СПИДа возможно рассматривать вопрос о прекращении антиретровирусной терапии.
Отмена иммуносупрессантов может привести к улучшению самочувствия пациентов. В то же время у этих пациентов повышается риск развития ВСВИС.
При развитии ПМЛ у пациентов, получающих лечение натализумабом, иным иммуномодулирующим лекарственным средством или иммуносупрессантами, прием указанных лекарственных средств необходимо прекратить, с последующим проведением плазмафереза для удаления остатков препарата в циркулирующей крови.
Ключевые моменты
ПМЛ развивается вследствие реактивации повсеместно распространенного JC-вируса, как правило, на фоне снижения клеточного иммунитета.
При ПМЛ чаще всего наблюдаются нарушение координации движений, гемипарезы, афазия, дизартрия, гемианопсия и когнитивные нарушения.
У пациентов со снижением клеточного иммунитета и необъяснимыми прогрессирующими признаками поражения головного мозга необходимы проведение МРТ головного мозга и исследование ЦСЖ на ДНК JC-вируса.
Проводите поддерживающую терапию пациентов и лечите основные заболевания согласно показаниям (например, прекратив прием натализумаба, другого иммуномодулирующего лекарственного средства или иммунодепрессантов, или инициируя антиретровирусную терапию и со всесторонним контролем за развитием воспалительного синдрома восстановления иммунитета).
Наследственные диффузный лейкоэнцефалопатия с сфероидов ( ЛВП ) является редким для взрослых Наступление аутосомно - доминантное заболевание характеризуется церебрального белого вещества дегенерации с демиелинизация и аксонов сфероида приводит к прогрессирующей когнитивной и моторной дисфункции. Сфероиды аксонов вздутие с прерывистым или отсутствием миелиновых оболочек. Считается , что заболевание возникает в результате первичной дисфункции микроглии , что приводит к дополнительному нарушению целостности аксонов, neuroaxonal повреждения и фокальных аксонов сфероидов , ведущих к демиелинизации . Сфероиды в HDLs напоминают до некоторой степени, продуцируемые напряжениями сдвига в закрытой черепно -мозговой травме с повреждением аксонов, заставляя их набухают вследствие закупорки аксонного транспорта . В дополнении к травме, аксоны сфероиды могут быть найдены в возрасте мозга, инсульте, а также в других дегенеративных заболеваниях. В ЛВПЕ, остается неясным , происходит ли демиелинизация до аксонов сфероидов или что вызывает нейродегенерацию после , по- видимому нормального мозга и белого вещества развития, хотя генетические дефициты позволяют предположить , что демиелинизация и аксоны патологии могут быть вторичными по отношению к микроглии дисфункции. Клинический синдром у больных с ЛВП не является специфичным и может быть ошибочно принята за болезнь Альцгеймера , лобно - височной деменции , атипичного паркинсонизма , рассеянный склероз , или кортикобазальной дегенерации .
содержание
Клинические симптомы
С симптомами изменения личности, поведенческих изменений, деменции , депрессии и эпилепсии, ЛВП был обычно диагностируется для целого ряда других заболеваний. Деменция или лобно поведенческие изменения, к примеру, обычно направляя некоторые клиницисты ошибочно считают диагнозы , такие как болезнь Альцгеймера, лобно - височная деменция или атипичного паркинсонизма. Наличие изменений белого вещества привело к неправильному диагнозу рассеянного склероза. ЛВП обычно проявляется с психоневрологическими симптомами, прогрессирующая деменцию, а через несколько лет показывает дисфункцию двигателя. В конце концов , пациенты становятся прикованными к инвалидной коляске или прикован к постели.
Белые дегенерации независимо от того , связаны с и делают дифференциальные диагнозы из других взрослых протекающих лейкодистрофия , такие как метахроматическая лейкодистрофия (MLD), болезнь Крабба (глобоидные клетки лейкодистрофия) и Х-хромосомы адренолейкодистрофия (X-ADL).
| болезнь | Эксклюзивный Тр |
|---|---|
| MLD | Накопление метахроматического материала в белом веществе |
| Краббе болезнь | Наличие глобоидных клеток, полученных из микроглии, которые имеют несколько ядер |
| X-ALD | Преобладающий теменно-затылочной белое вещество ненормальность |
| Исчезновение White Matter (VWM) Болезнь |
|
| Нас-Hakola |
|
Многие психоневрологические симптомы были выявлены в клинических исследованиях у больного ЛВПОМ. Они включают в себя тяжелую депрессию и тревогу , которые были определены в 70% от ЛВПА семей, доходящий суицидальных тенденций и токсикомании , таких как алкоголизм . Кроме того, пациенты могут проявлять дезориентацию, спутанность сознания, возбуждение, раздражительность, агрессивность, измененное психическое состояние, потеря способности выполнять выученные движения ( апраксия ), или неспособность говорить ( мутизм ).
Лица с ЛВПОМ могут страдать от толчков, снижение движения тела, нестационарность ( паркинсонизм , мышц на одной стороне тела в постоянном сжатии ( спастический гемипарез ), ухудшение в моторном и сенсорной функции нижних конечностей ( парапарез ), паралич приводит к частичному или полная потеря всех конечностей и туловища ( тетрапарез ), а также отсутствие добровольной координации движений мышц ( атаксия ).
Сопутствующие заболевания
Сопутствующие нарушения в том же спектре заболеваний , как HDLs включают болезнь Наса-Hakola ( поликистозный lipomembranous osteodysplasia с склерозирующей лейкоэнцефалопатией ), а также тип лейкодистрофия с пигментными заполненными макрофагами под названием пигментной ортохроматический лейкодистрофия (Põld). В дополнении к болезни белой материи, Нас-Hakola вызывает костные кисты. Это вызвано мутациями в генах , участвующих в одной и той же колониестимулирующий фактор (CSF) сигнального пути каскада , как определено в ЛВП.
Болезнь Нас-Хакол по- видимому, вызвана мутациями в Тайро белке тирозинкиназа-связывающий белке ( TYROBP - также известный как DAP12) или запускающего рецептор экспрессируется на миелоидные клетки (2 TREM2 ) белка. В то время как различные мутации гена происходят в пределах пути для Наса-Hakola и ЛВПА, и характеризуется дегенерацией белого вещества с аксонами сфероидов. Современные исследователи в этой области считают , что более глубоким анализом и сравнением двух генетических аномалий в этих нарушениях может привести к лучшему пониманию механизмов болезни в этих редких заболеваниях. PÕLD демонстрирует невоспалительные демиелинизации аксонов с начальными симптомами эйфории, апатия, головная боль и исполнительной дисфункции . В то время как ЛВП является аутосомно - доминантным, некоторые семьи с PÕLD имеют особенности , которые предполагают аутосомно рецессивное наследование. Тем не менее, PÕLD недавно было показано, что один и тот же генетический базис как ЛВП.
причины
Причиной HDLs в большинстве семей мутации в колониестимулирующий фактор 1 рецептор (CSF1R), фактор роста для микроглии и моноцитов / макрофагов, предполагая , что микроглии дисфункция может быть первичным в ЛВП.
Мутации сконцентрированы в тирозин киназы области (Таэквон) белка. Мутации в основном были найдены в экзонах 12-22 в внутриклеточном ТКДЕ, в том числе 10 миссенса мутация , которые имеют одну нуклеотидную делецию и один кодон удаления , который состоит из триплета нуклеотидов , которые были удалены в результате чего в целом аминокислоты , чтобы не быть закодирована. Кроме того, три сплайсинга мутации сайта были идентифицированы , что вызвало в рамке удаление из в экзоне , выраженной нуклеотидной последовательности, что приводит к удалению более чем 40 аминокислот в ТКД.
Это определение имеет на основании генетических исследований 14 HDLs семей , подтверждающих мутации в этом гене. Белка рецептора CSF1 в основном функции в регуляции, выживания, пролиферации и дифференциации клеток микроглии. Механизм микроглии дисфункции из - за мутаций в CSF1R к потере миелина и аксонов сфероида формирования остается неизвестным. Необходимы дальнейшие исследования , чтобы лучше понять заболевания патогенез .
патология
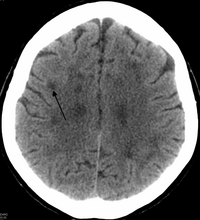
В ЛВП, есть расширение из боковых желудочков и отмечается истончение или ослабление мозговой белого вещества. Потеря белого вещества вызвано миелина потери. Эти изменения связаны с диффузным глиозом , умеренной потерей аксонов и многих аксонов эллипсоидов.
Активированный или амебоидная микроглия и макрофаги , которые содержат миелин мусора, липидные капли и коричневые гранулы пигмента autofluorescent встречаются в районах с демиелинизацией и аксональными сфероидами. В сильно вырожденных областях есть много больших, реактивные астроциты , наполненные глиальных фибрилл .
В случаях аутопсии, было показано , что белое вещество аномалия относительно ограничена в головной мозг , избегая при этом мозжечке , и многие из основных волоконных участков нервной системы. Исключение составляет корковы массивы (пирамидальные трактов) в стволе головного мозга , а иногда и спинной мозг .
Патология мозга HDLs напоминает болезнь НАНЫ Hakola (поликистозного lipomembranous osteodysplasia с склерозирующей лейкоэнцефалопатией).
диагностика
Исследования в 2012 включает в себя исследование микроглии функции. Эта работа будет способствовать дальнейшему выяснить, является ли эта болезнь в основном дефект функции микроглии. Для такого исследования, клетки микроглии из HDLs родственных можно культивировать из мозга аутопсии и анализировали в сравнении с нормальными клетками микроглии на основе различий в мутационных событий и экспрессии фактора роста.
Для того, чтобы получить лучшее понимание болезни, исследователи ретроспективно проанализировали медицинские записи пробандов и другим , которые были оценены с помощью клинических исследований или вопросников. Образцы крови собирают из семей пробандов для генетического тестирования. Эти члены семьи оценивают по их стандартную медицинскую историю , на их прогрессирование как симптомов болезни Паркинсона ( Унифицированная оценки болезни Паркинсона Scale ), и на их прогрессирования когнитивных нарушений , таких как деменция ( Folstein Test ).
Стандартный МРТ сканирование было выполнено на 1,5 Тесла сканеров с толщиной 5 мм и шагом 5 мм для скрининга белого вещества поражений в определенных семьях. Если интенсивность сигнала в МРТ в белых районах вещества выше , чем в серых областях материи, пациент считаются риском ЛВПА, хотя ряд других заболеваний , также может производить белые изменения материи и выводы не являются диагностическими без генетического тестирования или патологическое подтверждение.
Тканевые срезы биопсии головного мозга или головного мозга аутопсии , как правило , встроен в парафин , из которого секции вырезать смонтированы на предметные стекла для гистологических исследований. Окрашивание для миелина и аксонов патологии показывают патологические изменения, характерные для ЛВП определены в белом веществе неокортекса , базальных ганглиев , таламуса , среднего мозга , мосте и спинного мозга. В дополнение к обычным гистологических методов ( Н & Е окрашивания ), образцы оценивали с иммуногистохимии для убиквитина , амилоидного белка - предшественника, и нейрофиламентов , чтобы охарактеризовать аксонов изменения и основной белок миелина для миелина патологии. Иммуногистохимических пятна для микроглии (CD68 или HLA-DR) и астроцитов (GFAP), также полезные методы для характеристики белого вещества патологии. С аналогичной патологией в PÕLD, ЛВП обычно группируются во взрослом возрасте лейкоэнцефалопатии с аксонами эллипсоидов и пигментированной глией (ALSP), чтобы дать этим отдельно в соответствии с признанными условиями повышенного внимания.
ЛВП подпадает под категорию мозга заболеваний белого вещества под названием leukoencephalopathies, которые характеризуются определенной степенью дисфункции белого вещества. ЛВП имеет повреждения белого вещества с нарушениями в миелиновой оболочки вокруг аксонов, где причинные влияния в настоящее время постоянно проводятся исследования на основе последних генетических исследований. Исследования Sundal и коллегами из Швеции показали , что аллель риски кавказцев может быть причинной , поскольку случаи , выявленные до сих пор были среди крупных кавказских семей.
управление
эпидемиология
Средний клинический профиль из опубликованных исследований показывает, что средний возраст начала для пациентов ЛВПА это 44,3 лет со средней длительностью заболевания 5,8 года и средним возраст смерти в 53,2 лет. По состоянию на 2012 год, там было около 15 случаев, идентифицированные по крайней мере, 11 спорадических случаев ЛВП. случаи ЛВПОВ были расположены в Германии, Норвегии, Швеции и США, демонстрируя международное распространение фокусирующего между Северной Европой и Соединенными Штатами.
На основе изучения многочисленных родстве, было установлено , что заболевание не происходит среди всего мужчин и женщин, а равномерно распределены указывает на аутосомно , а не сцепленных с полом генетическим расстройством . Было также отмечено , что случаи ЛВП не пропустить поколений , как это будет происходить с рецессивным наследованием, и как таковой был назван аутосомно - доминантным.
история
Эта болезнь впервые была описана в 1984 г. Аксельсон и соавт. в большой шведской родословной. Это расстройство более известный невропатологов , чем врачи. Невропатолог с интересом ЛВП, д - р Деннис У. Диксона, был выявлен ряд случаев из невропатологии исследования головного мозга , представленных для исследования семейных взрослого начала деменцией и двигательных расстройств в Нью - Йорке , а затем во Флориде. Признание важности этого заболевания в качестве причины для взрослых протекающей деменции и двигательных расстройств дополнительно усиливается в 1997 году в клинике Майо , когда Збигнев К. Wszolek определила семью с ЛВП, первоначально считались, что из - за другой болезненный процесс ( FTDP-17), но только вскрытие трупа одного и затем других членов семьи показало , что это будет ЛВП. Wszolek создан международный консорциум , в 2005 году для выявления других семей и собрать ДНК или мозговые образцы из членов семьи для подтверждения нейропатологического и генетических исследований в клинике Майо в штате Флорида.
Читайте также:
